Article Text

Abstract
Interstitial lung disease in children (chILD) is rare, and most centres will only see a few cases/year. There are numerous possible underlying diagnoses, with specific and non-specific treatment possibilities. The chILD-EU collaboration has brought together centres from across Europe to advance understanding of these considerations, and as part of this process, has created standard operating procedures and protocols for the investigation of chILD. Where established consensus documents exist already, for example, for the performance of bronchoalveolar lavage and processing of lung biopsies, these have been adopted. This manuscript reports our proposals for a staged investigation of chILD, starting from when the condition is suspected to defining the diagnosis, using pathways dependent on the clinical condition and the degree of illness of the child. These include the performance of genetic testing, echocardiography, high-resolution CT, bronchoscopy when appropriate and the definitive investigation of lung biopsy, in order to establish a precise diagnosis. Since no randomised controlled trials of treatment have ever been performed, we also report a Delphi consensus process to try to harmonise treatment protocols such as the use of intravenous and oral corticosteroids, and add-on therapies such as hydroxychloroquine and azithromycin. The aim is not to dictate to clinicians when a therapeutic trial should be performed, but to offer the possibility to collaborators of having a unified approach when a decision to treat has been made.
- ABCA3
- Idiopathic pulmonary fibrosis
- Surfactant protein
- TTF-1
- PIG
- NEHI
- GM-CSF Receptor
- Hypersensitivity pneumonitis
Statistics from Altmetric.com
- ABCA3
- Idiopathic pulmonary fibrosis
- Surfactant protein
- TTF-1
- PIG
- NEHI
- GM-CSF Receptor
- Hypersensitivity pneumonitis
Introduction
Interstitial lung disease in children (chILD) is rare; there are no reliable estimates, but prevalence is likely <1 per 100 000, as against 60–80 per 100 000 in adults;1 the average European Hospital will see no more than 5 cases/year.2 chILD is more diverse than interstitial lung disease in adults, comprising more than 200 different conditions with a variety of proposed classifications.3–6 Since individual centres see so few chILD patients, it is unsurprising that there are no randomised controlled trials of treatment. To address this, a proposal was funded by the European Union FP7 stream7 to (a) create a pan-European registry; (b) peer review all potential chILD diagnoses; (c) gather prospective longitudinal data from well-defined groups of children, including the results of N of 1 therapeutic trials; (d) set up and perform the first randomised controlled trials of chILD treatment. In order to achieve this, it was essential that the diagnostic approaches and treatment protocols were harmonised across Europe. This manuscript reports on our agreed approach, including standard operating protocols, and the Delphi consensus process used to harmonise our approach to treatment, across the entire childhood age range. The reader is also referred to an official American Thoracic Society statement, which covers only the 0–2 years age range.8 The aim is not to dictate to clinicians whether to undertake trials of particular treatments but to encourage and enable the use of the same protocol for treatments where there is no evidence.
The starting point: when to suspect ChILD—typical presentations
Shortly after birth
The earliest presentation of chILD is shortly after birth, with unexplained respiratory distress in a term baby. This frequently and rapidly proceeds to intubation and ventilation, with relentlessly progressive respiratory failure8 ,9 leading to death or lung transplantation, if the latter is available. The usual causes are surfactant protein gene mutations9 ,10 and the alveolar-capillary dysplasia spectrum;11 some of the former may make a partial recovery10 and the baby may survive in chronic respiratory failure. The same conditions should be considered in preterm babies with respiratory distress which does not run the normal clinical course, but the yield of positive results is much less.
First 2 years of life
Later presentations of chILD are very non-specific. In the first 2 years of life, symptoms in descending order of frequency are fast breathing, failure to thrive, typically dry cough and wheeze (this last in 25% of cases) in the absence of respiratory tract infection.3 Suggested protocols for the clinical assessment of these infants are available on the chILD-EU website (http://www.klinikum.uni-muenchen.de/Child-EU/en/index.html). SpC mutations may present as respiratory syncytial virus (RSV)-positive bronchiolitis, which fails to resolve;12 there is animal evidence that RSV causes increased inflammation and worse outcomes in animals with SpC mutations.13 ,14 Signs of chILD include hypoxia, tachypnoea, recession, crackles, pulmonary hypertension and gastro-oesophageal reflux.3 Auscultation can be normal in more than a third. Chest deformity, particularly pectus excavatum, has been reported especially with ABCA3 mutations.15
2–16 years of age
There are no large series from this age group, but it is likely that the clinical picture will not be dissimilar. In older children, exercise-induced breathlessness may be the presentation, although other, commoner causes need to be considered.
The USA group have coined the term ‘chILD syndrome’7 which is a label mandating further evaluation in a child with diffuse lung disease who has had common causes excluded (eg, cystic fibrosis, uncoordinated swallow leading to aspiration) and who has at least three of four criteria, namely (a) respiratory symptoms; (b) respiratory signs, including digital clubbing and failure to thrive; (c) hypoxaemia; and (d) diffuse radiological changes. Clearly, another important part of the initial work-up is a full clinical evaluation of the child, looking for signs of a systemic disease (especially renal, skin, liver, eyes, joints), evidence of pulmonary hypertension and signs of reflux and aspiration.
In all cases, a careful history is mandated, including a family history, which may lead to suspicion of genetic causes of chILD, and environmental exposures if there is any possibility of hypersensitivity pneumonitis; the recent outbreak of ILD in adults and children in Korea emphasises the importance of the environmental history.16–18 Simple laboratory testing may give clues to the diagnosis. Anaemia and reticulocytosis are seen in pulmonary haemorrhagic syndromes. Peripheral eosinophilia suggests parasitic disease, hypersensitivity, eosinophilic lung disease and systemic eosinophilic diseases. Stool occult blood results may be positive in patients with pulmonary haemorrhage; indeed these children may have undergone extensive diagnostic work-up for iron deficiency without the possibility of pulmonary haemorrhage having been considered.19
Initial investigations
Given the non-specific nature of the presentation of chILD the probability of a diagnosis can be enhanced by structured investigation. A simple severity score has been proposed (table 1),20 which sadly has yet to be superseded by more sophisticated biomarkers:
Clinical: Evidence of hypoxaemia is often the first abnormality to raise concern and should be noted whether it is present at rest both awake and asleep breathing room air. In older children exercise testing should be used to unmask desaturation which is not present at rest, following standard protocols; there is insufficient evidence to recommend a particular method over others. It is unwise and uninformative to exercise a child who is desaturated at rest, without giving supplemental oxygen.
Radiology: The chest radiograph may be normal, but more likely reveals non-specific abnormalities. Gastro-oesophageal reflux is common in young children and also in patients with respiratory disease; deciding if reflux may be contributory is difficult even in the presence of a barium swallow reflux study and a pH study.
Physiological testing: Lung function testing in infants has little general availability and the role in infants is unclear. Neuroendocrine cell hyperplasia of infancy (NEHI) is characterised by a pattern of hyperinflation and airflow obstruction,21 but there is insufficient experience to rely on physiological patterns in infancy in chILD. In older children, the usual pattern is classically restrictive, with a reduced FEV1 and FVC, a normal or even elevated FEV1/FVC ratio and reduced lung volumes. Carbon monoxide transfer (diffusing capacity or transfer factor of the lung for carbon monoxide, DLCO) is usually reduced, and if elevated, pulmonary haemorrhage should be suspected.22 The occasional chILD may have a more obstructive pattern of physiology.
Echocardiogram: Should be an early investigation to estimate pulmonary artery pressure, and exclude cardiac mimics of interstitial lung disease, such as cor triatrium leading to pulmonary oedema.23 Pulmonary hypertension, where present, should be diagnosed and treated accordingly in liaison with paediatric cardiology colleagues.
Blood studies: A focused approach is recommended. Tests can be grouped into (a) genetic abnormalities; (b) immune function (especially if the follicular bronchiolitis-lymphoid interstitial pneumonia spectrum is suspected); (c) auto-antibody studies (cases of pulmonary haemorrhage, alveolar proteinosis, or if there is evidence of a systemic disease); (d) environmental organic dust exposures (hypersensitivity pneumonitis); (e) miscellaneous, that is, ACE inhibitors in cases of suspected sarcoidosis. Note that novel genetic chILD entities with multisystem disease are being described,24 ,25 and the younger the child is the more carefully new or established genetic diagnoses are sought. In all cases of chILD, DNA of the patient and parents should be stored for future analyses. The clinical situation will dictate which tests are performed, and whether it is realistic to await results before proceeding to a CT or lung biopsy.
A proposed diagnostic pathway is shown in figure 1. In terms of timing of enrolment of suspected chILD patients in the registry, a preliminary discussion at an early stage would be valuable for the patient. The use of, for example, standardised scanning protocols is to be preferred.
Staging the severity of childhood interstitial lung disease (chILD)20
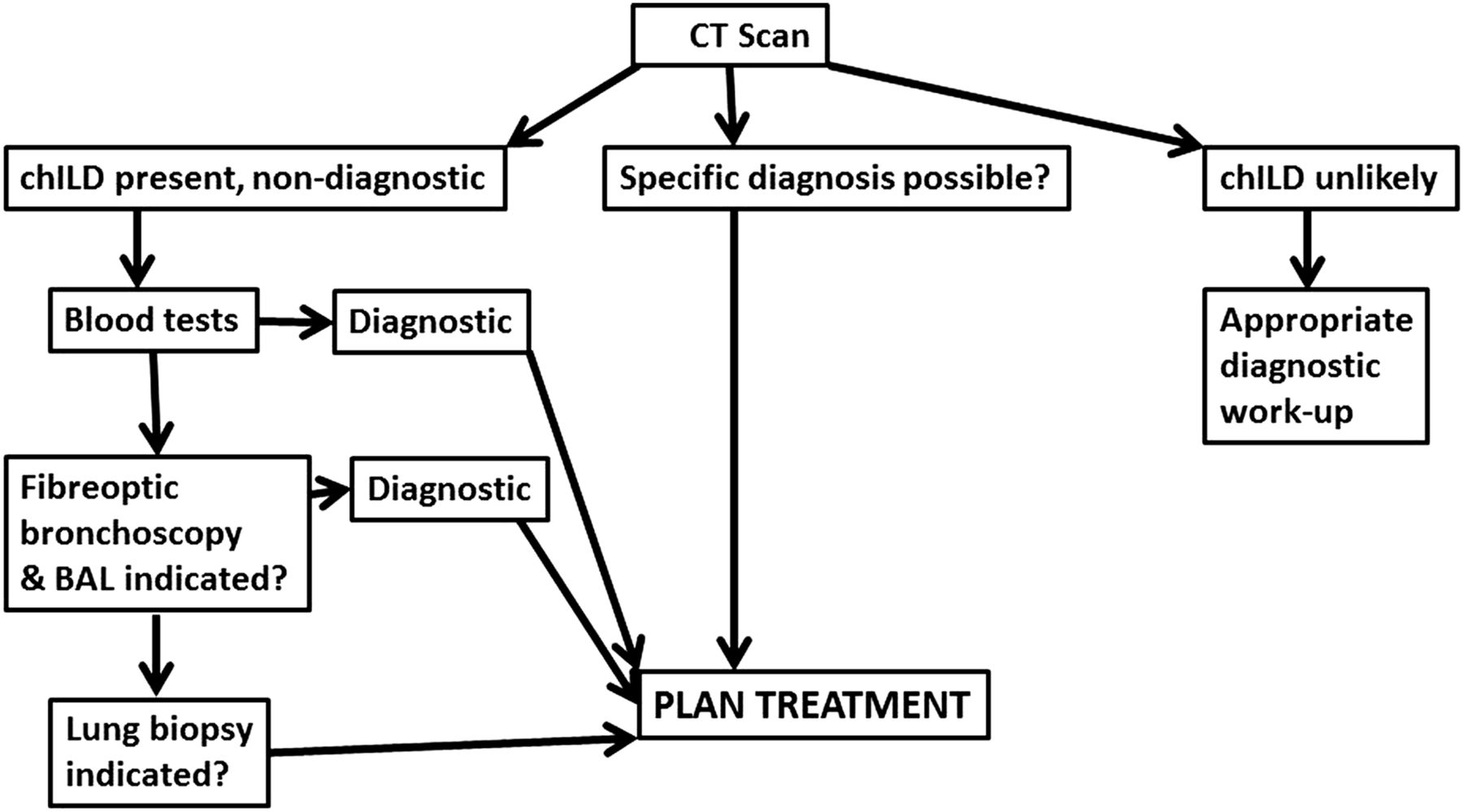
Proposed flow chart for investigating interstitial lung disease in children (chILD). If the child is well and stable, progress to the more invasive investigations may not be indicated. HRCT, high-resolution CT.
If chILD is suspected clinically: the role of chest CT scanning
CT chest is invariably performed in the investigation of a possible case of ChILD. The purpose of chest CT is to evaluate the presence and extent of disease. In some cases it can be diagnostic,26 but is more usually supportive of a diagnosis that takes account of clinical history, blood results and sometimes bronchoalveolar lavage and/or biopsy material.
CT scanning for chILD should only be performed in centres experienced in paediatric radiology. The general principles of scanning are to minimise radiation dosage to the child while maximising the information obtained; a very low dose scan which is diagnostically useless benefits no one.27 Detailed protocols for reducing radiation exposure while maintaining image quality are given online, and these should be audited regularly.
The optimal CT scan is a volumetric scan during inspiration, performed in tandem with a high-resolution CT (HRCT) fine-cut spaced expiratory scan. Ventilation should be controlled to ensure satisfactory, interpretable, scan output. In young children this necessitates a general anaesthetic (ie, under 5 years of age), although some centres advocate sedation and controlled, bag and mask ventilation.28 As always, the risk of anaesthesia or sedation should be weighed against the risk of not obtaining diagnostic information. Older children should be able to reliably hold breath in inspiration and expiration for 6–10 s. In children who are ventilated under a general anaesthetic it is important to attain appropriate ventilation pressures in both inspiration and expiration; these details are provided in our online standard operating procedure (SOP).
The administration of contrast medium will make the assessment of ground glass shadowing almost impossible, and so careful consideration should be given as to the risk/benefit of using a contrast medium based on the anticipated diagnosis. The need to assess the pulmonary vasculature will require the use of contrast, with detrimental effects on the interpretation of some lung components.
The three outcomes of HRCT scanning (figure 1) are (1) chILD unlikely including that the CT is normal. Although a normal CT scan is unusual in ChILD, it may occur in some cases early in the course of the disease process where there are very few non-diagnostic CT features; (2) chILD present and a specific diagnosis can be made26; and (3) chILD present, with or without possible evidence of a comorbidity such as aspiration (eg, consolidation in a typical distribution), but a specific diagnosis cannot be made. If chILD is unlikely then appropriate diagnostic algorithms to investigate possible non-chILD findings are deployed; these are beyond the scope of this article. A specific diagnosis of chILD may allow immediate action; however, the ability for CT to be completely diagnostic is limited to relatively few conditions (ie, pulmonary alveolar proteinosis, extrinsic allergic alveolitis and NEHI), and these too are usually supported by other diagnostic tests, that is, environmental allergen and prednisolone response in hypersensitivity pneumonitis (figure 2A) ,or granulocyte-macrophage colony-stimulating factorreceptor auto-antibody or gene mutation studies, surfactant protein gene mutations and an immunological work-up in cases of pulmonary alveolar proteinosis (figure 2B).

{kind=link}
{kind=link}
Diagnostic high-resolution CTs (HRCTs) in childhood interstitial lung disease (chILD). (A) There is diffuse ground glass shadowing with a widespread soft centrilobular nodular pattern. This is virtually diagnostic of hypersensitivity pneumonitis, in this case due to pet doves. (B) There is generalised ground glass opacification with thickening of the interlobular septa, giving the classical cobblestone appearance of pulmonary alveolar proteinosis. It is not possible to distinguish the different aetiologies from the HRCT appearances.
It is recognised that although faster CT scanners may enable chest CT without the need for anaesthetic, we do not recommend this unless anaesthesia is thought to be unsafe, as (1) the technique frequently provides suboptimal results from respiratory movements made by the child and (2) the variance in lung volumes during an uncontrolled respiratory cycle will reduce the value of the scans obtained and the reliability of CT to provide the supportive evidence for a diagnosis of ChILD.
Whatever the timing of investigations, planning to avoid the child having multiple general anaesthetics is essential. So fibre-optic bronchoscopy should only be done as a separate procedure if it is likely to be diagnostic; otherwise, it is better performed under the same anaesthetic as the lung biopsy.
Invasive testing: flexible bronchoscopy
Bronchoscopy with bronchoalveolar lavage (BAL) may be diagnostic in pulmonary haemorrhage syndromes, alveolar proteinosis and eosinophilic lung disease, and a normal cell differential can rule out hypersensitivity pneumonitis. BAL target site should be chosen on the basis of radiological abnormality or airway inspection. If BAL is to be performed during the same anaesthetic as CT, then it should follow the imaging; where it is to be performed at the time of lung biopsy, the lobe designated for a lung biopsy should be avoided. Where possible flexible bronchoscopy should be performed via endotracheal or laryngeal mask to reduce suction channel contamination from the upper airway. BAL volume is adjusted to body weight using 3 mL/kg of sterile normal saline divided into three equal fractions in children weighing <20 kg and 3 mL/kg in 20 mL portions in children weighing >20 kg.29 Negative suction pressure should be adjusted to avoid visible airway collapse. The first bronchoalveolar lavage fluid aliquot should be unfiltered and used for microbiological studies, and the other aliquots should be pooled (unless one is bloodstained), filtered through sterile gauze only if a lot of mucus is present, which is unlikely in chILD, and used for analysis of cellular and non-cellular components. Transport and preparation of samples is provided in our online SOP. Clinicians should liaise with their laboratories to ensure appropriate investigation of samples, to include appropriate microbiological analysis and culture (bacterial, fungal, virus), Oil Red O staining (fat-laden macrophages), periodic acid–Schiff staining (pulmonary alveolar proteinosis) and iron staining (ie, Prussian blue for haemosiderin within macrophages) to diagnose pulmonary haemorrhage syndrome. CD1a-positive cells are consistent with Langerhans cell histiocytosis. A lymphocyte-predominant BAL with an elevated CD4/CD8 ratio (>2) suggests sarcoidosis with involvement of the lung.30 Metabolic abnormalities may be diagnosed by BAL, for example, sea-blue macrophages.31
Endobronchial (EBx) and transbronchial (TBB) biopsies are rarely performed in ChILD and are not recommended unless a specific diagnosis that could be made by these techniques is suspected. Specific conditions such as pulmonary alveolar microlithiasis may be diagnosed by TBB,32 but the samples are too small to diagnose most chILDs. Blind EBx may detect sarcoid granulomas,33 but is not useful for diagnosing NEHI.34 Our protocols for these procedures can be found online if needed.
Invasive testing: surgical lung biopsy
The timing and need for lung biopsy is controversial. chILD patients who are well and thriving may not merit biopsy even if the CT scan appearances are not typical. Some would consider that an oxygen requirement warrants a diagnostic lung biopsy, while others would wait and see. There is clearly merit in performing an invasive procedure only if treatment will be changed as a result. Steroids are a mainstay of treatment for chILD and the timing of biopsy related to their initiation is often dictated by circumstance. Where possible, biopsy prior to steroid treatment is recommended (to minimise risk to wound healing and to expedite specific chILD treatments, ie, TNF-α antagonist infliximab combined with methotrexate for sarcoidosis, and cyclophosphamide for angiitis with granulomatosis). The dilemma posed by the sick patient with chILD who is on the verge of ventilation (and biopsy would most likely tip to requiring ventilation) or is unstable on a ventilator often dictates that a steroid trial before a biopsy may be appropriate. If the child is already ventilated, unless the ventilatory requirements are very high, a biopsy can safely be performed.
The site of biopsy should be guided by a recent CT chest. There should be liaison between the surgeon, pathologist and paediatrician. Any other procedures which may merit general anaesthesia (eg, bronchoalveolar lavage, mucosal biopsy, placement of a vascular access device or gastrostomy) should be carefully planned. The tip of the middle lobe and lingula should be avoided, and biopsy should preferably be from two sites, and sample areas of varying disease severity. Increasingly, biopsy is using video-assisted thoracoscopic surgery rather than a minithoracotomy. Whatever technique is used, the procedure must only be undertaken by an experienced surgeon, who is confident of obtaining adequate biopsies (at least 10×10×10 mm); a very superficial biopsy, which does not contain distal airways, may result in diagnostic error.
Samples should be rapidly transported to a waiting pathologist working in an accredited laboratory working to international standards.35 There should be access to light microscopy and common special stains, immunohistochemistry, genetics services, microbiology and virology services and photographic equipment. Table 2 describes the postbiopsy sample processing. The remaining tissue should be fixed in formalin. Table 3 describes the routine stains to be used. Details of transport of specimens can be found online.
Handling of lung biopsy specimens
Staining of lung biopsy specimens
Other sources of lung tissue
Postmortem and explanted lung tissue should be accessed if at all possible, and handled in the same way as a lung biopsy. Even if permission for autopsy is refused, it may be possible to obtain permission for percutaneous, transthoracic True-cut needle biopsies. DNA of the patient and both parents should be stored with the family's permission, if this has not already been done. Another valuable source of tissue is the explanted lungs after lung transplantation.
The role of peer review
chILD is a spectrum of very rare conditions, and no centre will see many cases. The diagnosis may be obvious, as, for example, a suspected surfactant protein gene mutation when known disease causing mutations are detected. However, in many cases the interpretation of CT scans and lung biopsy material is difficult. We believe that the best practice is to peer review these cases so that patients have the benefit of a consensus view from different specialists who can pool and increase their expertise. This is an integral part of our protocols. In chILD-EU, cases are peer reviewed at point of diagnosis and revisited at 12 monthly intervals to gauge the precision of diagnosis and patient outcome.
Proposed treatment protocols: the Delphi process
In the absence of any randomised controlled trial data, a Delphi process was undertaken to achieve a consensus on treatment protocols to harmonise treatment approaches. The full details are given in the online supplementary material. Briefly, we conducted a two-round web-based Delphi consensus of clinicians caring for chILD. Clinicians were identified from registered email addresses with national paediatric respiratory society groups, including the paediatric assembly of the American Thoracic Society. The questions were derived by the chILD-EU core management team, with adaptation by the lead representatives for clinicians in North America (Robin Deterding) and Australia (Adam Jaffe). The survey consisted of a scenario stem and related questions. All scenarios related to children with proven or highly suspected ILD. The survey was in English. Survey 1 consisted of 8 scenario stems with 75 responses required, survey 2 of 9 lead statements with 11 individual responses required. Clinicians were asked to score according to agreement: 1 Strongly Agree; 2 Agree; 3 Neutral; 4 Disagree; 5 Strongly Disagree; 6 Don't know.
Survey 1 was approached by 173 clinicians. Twenty-nine responders who provided fewer than 50% of responses were removed from the analysis. Data is presented from 144 clinician responders: Australia 14, Canada 3, France 5, Germany 15, Italy 1, USA 61, UK 37 and other 8. There were 127 respondents to the second round of Delphi. Twelve provided insufficient data responses (fewer than 50% responses) and so were removed from the complete analysis. The 115 consultant/attending pulmonologist respondents contributing to the second round of Delphi were from the UK (36), USA (30), Germany (22), Australia (9), France (6), Italy (5), Canada (2) and other countries (5).
Delphi 1 consisted of four scenarios related to the care of a patient who was very sick and ventilated or close to ventilation for whom no specific treatment was known. Delphi 2 provided statements derived from the consensus of opinion from the first Delphi round and asked how clinicians viewed the statement. Options, provided for varying doses and medicines, are summarised as (a) usual practice or (b) could happily use it if it is the consensus of the Delphi, or (c) not usual practice and would not wish to use it even if it is the consensus of the Delphi.
In all scenarios, we were clear that the suggested doses should be considered starting doses and could be changed at the discretion of the treating clinician at any time. The final consensus for treatment reached is summarised in table 4, and the consensus for a significant response in table 5.
Doses of medication to be used and anticipated time to assess if there has been a clinical response
Improvement in physiological outcomes considered to be a treatment response
Other potential therapies
There is even less evidence for other regimes and no recommendations are made. If there is evidence of systemic disease, and if cytotoxic or biological therapies are contemplated, it would usually be wise to involve a paediatric rheumatologist.
Monitoring chILD
The variety of ChILD diagnoses makes a single common monitoring plan of little value. chILD-EU has looked to enable reference across diagnoses by the development of an observational trial protocol focused on the first year of diagnosis. Monitoring is at months 1, 2, 3, 6 and 12 and annually thereafter. Key observations are clinical (respiratory rate, heart rate, weight, oxygen saturation in air awake, oxygen saturation including overnight while asleep and on exercise, evidence of pulmonary hypertension) and radiological monitoring (chest X-ray at diagnosis, 6 and 12 months; CT not recommended, however, if considered justifiable, a limited cut thin-section HRCT of areas of interest should provide sufficient information). In older children spirometry at each observational monitoring visit should be recorded, with DLCO (pulmonary haemorrhage monitoring) and body plethysmography recommended as indicated but at least once per year.
Pulmonary exacerbations
Acute pulmonary events associated with instability are a characteristic of many chronic diseases. Although recognised, they are often difficult to define. No definition exists for a pulmonary exacerbation in chILD and so we have sought, through the Delphi process and a review of the evidence, to define what is a clinically significant change in chILD as a first step towards a definition of a pulmonary exacerbation (table 5). The period of stability before and between exacerbations must be defined; for ChILD this is arbitrarily considered to be at least 7 days of stability at baseline values. More work is ongoing to define the definition of a pulmonary exacerbation and its relationship with other respiratory events such as upper respiratory tract infection and aspiration.
Summary and conclusions
We have proposed a sequence of investigations for chILD, and provided SOPs for performance of these tests. We aim to harmonise investigation of chILD to facilitate future research; this manuscript is the framework on which this will be based.
Acknowledgments
We are grateful to Professor Robin Deterding, ChILD Foundation and Children's Hospital Colorado, USA, and Professor Adam Jaffe, Sydney Children's Hospital, Australia, for their assistance co-ordinating national responses via their respective networks (American Thoracic Society Paediatric Section and Australian Paediatric Medicine Respiratory Group).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Contributors AB drafted the manuscript, all authors reviewed it and agreed on the contents. SC led the Delphi consensus, and wrote the online supplement. All authors reviewed it and agreed with the contents.
Funding This project (“Orphans Unite: chILD better together – European Management Platform for Childhood Interstitial Lung Disease”) was funded by the FP7-305653-chILD-EU grant. In addition, AB and AGN were supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London.
Competing interests None declared.
Provenance and peer review Not commissioned; internally peer reviewed.
Data sharing statement Original data from the Delphi consensus available through SC.
Linked Articles
- Airwaves