Article Text

Abstract
Background Airway epithelial cells (AEC) from patients with asthma, appear to have an impaired interferon (IFN)-β and -λ response to infection with rhinovirus.
Objectives To determine if impaired IFN responses can be identified in young children at risk of developing asthma due to atopy and/or early life wheeze, and if the site of infection or the infecting virus influence the antiviral response.
Methods Nasal (N) and tracheal (T) epithelial cells (EC) were collected from children categorised with atopy and/or wheeze based on specific IgE to locally common aeroallergens and a questionnaire concerning respiratory health. Submerged primary cultures were infected with respiratory syncytial virus (RSV) or human metapneumovirus (hMPV), and IFN production, inflammatory cytokine expression and viral replication quantified.
Results Nasal epithelial cells (NEC), but not tracheal epithelial cells (TEC), from children with wheeze and/or atopy produced less IFN-β, but not IFN-λ, in response to RSV infection; this was associated with higher viral shedding. However, IFN-regulated factors IRF-7, Mx-1 and CXCL-10, and inflammatory cytokines were not differentially regulated. NECs and TECs from children with wheeze and/or atopy demonstrated no impairment of the IFN response (β or λ) to hMPV infection. Despite this, more hMPV was shed from these cells.
Conclusions AECs from children with wheeze and/or atopy do not have an intrinsic defect in the production of IFN-β or -λ, however, this response is influenced by the infecting virus. Higher viral load is associated with atopy and wheeze suggesting an impaired antiviral response to RSV and hMPV that is not influenced by production of IFNs.
- Airway Epithelium
- Asthma
- Asthma Mechanisms
- Innate Immunity
- Respiratory Infection
- Viral infection
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Key messages
What is the key question?
-
Are airway epithelial cells from the upper and lower respiratory tract of young children at risk of developing asthma, based on recurrent wheeze and/or atopy intrinsically deficient in the innate immune response to infection by the common childhood viruses respiratory syncytial virus and human metapneumovirus?
What is the bottom line?
-
Wheeze and atopy in young children are associated with a defect in the antiviral response of airway epithelial cells that is not dependent on interferon (IFN) production, results in elevated viral replication, and is influenced by the infecting virus.
Why read on?
-
Wheeze and atopy in young children at risk of developing asthma are associated with defective antiviral responses that are regulated by the infecting virus and are not strongly influenced by IFN production.
Introduction
Major risk factors for the development of asthma are a family history of asthma/atopy, severe respiratory viral infections associated with wheeze and/or fever in early life, and early sensitisation to aeroallergens (atopy).1–3 Respiratory viral infections are the principal cause of asthma exacerbations in children,4–6 with rhinovirus (RV) being the most commonly detected.7 However, frequent wheezing episodes early in life caused by other respiratory viruses, such as the paramyxoviruses respiratory syncytial virus (RSV) and human metapneumovirus (hMPV), have been linked with the later development of asthma; particularly RSV-induced bronchiolitis in infancy.1 ,8
An impaired innate immune response to RV infection, has been reported in adults with asthma, and has been proposed to increase susceptibility to viral infections. Infection of primary airway epithelial cell (AEC) cultures demonstrated that asthma-affected AECs produced less interferon (IFN)-β9 and -λ,10 in response to RV. However, not all studies have shown an impaired IFN response by adults with asthma.11 ,12 Additionally, few studies have investigated antiviral immunity to respiratory viruses other than RV; so whether this apparent deficiency in the IFN response represents an intrinsic deficiency to all respiratory viruses is not known.
Atopy is a major risk factor associated with the development of asthma. Specifically, ex vivo infection of AECs with RV, and biopsy specimens taken from children with atopy and/or asthma, demonstrated that a deficient IFN response to RV infection was equally present in atopic and non-atopic patients with asthma and atopics without asthma.13
The present study was undertaken to determine the potential for intrinsic defects in the antiviral response to be present in AECs from children who are not diagnosed with asthma, but are at risk of developing asthma later in life based on allergic sensitisation and a history of early life viral wheeze. We infected nasal (N) and tracheal (T) epithelial cells (ECs) ex vivo with RSV and hMPV to investigate the antiviral response, and also to determine if responses differed with the infecting virus, and between the upper and lower airways.
Methods
Subjects and sample collection
Children, aged 2–10 years, scheduled for elective otolaryngological surgery, primarily for adenotonsillectomy, necessitating intubation, were recruited (table 1). Parents answered a standardised questionnaire about the child's respiratory health, family history and environmental exposures in the home. Children were categorised as having a history of viral wheeze based on doctor diagnosis, frequency of visits to a general practitioner, emergency departments, or admission to hospital. None of the children included in the study were diagnosed with asthma, or prescribed anti-inflammatory therapy. Allergen-specific IgE to locally common aeroallergens (house dust mite, mixed grasses, animal hair, and mould) were measured (Immunocap, Thermo Scientific) and allergic sensitisation was defined as IgE≥0.35 kU/L to one or more aeroallergen. Children were classified into four mutually exclusive groups: healthy; atopy no wheeze (atopy alone); wheeze no atopy (wheeze alone); wheeze and atopy (table 1). All children were healthy (no rhinitis) at the time of sampling. Following induction of general anaesthesia, but prior to commencement of surgery, nasal scrapings were obtained from the inferior turbinate of each nostril using a purpose-designed curette (ASI Rhino-Pro, Arlington Scientific, USA) and tracheal brushings obtained by passing a cytology brush (0.5 cm brush at the end of a guide wire with bristles 1–2 mm long) through the tracheal tube. Peripheral blood was collected by venipuncture.
Demographic data for subjects recruited to study
Submerged culture of primary epithelial cells
Primary cultures were established by seeding freshly collected nasal epithelial cells (NEC) or tracheal epithelial cells (TEC) into steroid supplemented bronchial epithelial growth medium (BEGM; Lonza) in 25 cm2 cell culture flasks. Ninety percent of these cultures survived to be seeded at passage 2 into 12-well plates, and cultured until 75% confluent (63% cultures survived to this density). The culture medium was then replaced with steroid-free medium 24 h prior to infection with RSV or hMPV.
Infection with RSV or hMPV
Primary cells were exposed to virus at a high multiplicity of infection (MOI) of either three plaque-forming units (pfu)/cell (for RSV; A2) or three times the dose required to infect 50% of cells in culture (TCID50 units)/cell (for hMPV; CAN97-83) or low MOI of 0.1 pfu (or TCID50 units)/cell, to investigate the kinetics of infection over time. Uninfected control replicates were exposed to cell culture medium. Cells were incubated with virus for 2 h at 37°C, after which time they were washed three times with 1×PBS and the media replaced with steroid-free culture medium. Details of viral preparation are given in the online supplementary information.
Quantification of viral replication
Viral replication was determined by measuring the amount of live virus shed into the culture supernatant (MOI 0.1), or by quantitative PCR of intracellular viral RNA (MOI 3). Culture supernatants of AECs infected with RSV or hMPV at a MOI of 0.1 were collected at days 0, 1, 3 and 5 postinfection (D0–D5). Only those cultures that survived for the full 5 days were sampled. Supernatants were used in a plaque assay14 for quantification of RSV, or a TCID50 assay15 for quantification of hMPV (see online supplementary information). RNA was extracted from cells infected at a MOI of 3, and RT-qPCR performed to quantify intracellular viral RNA (see online supplementary information).
Cytokine and IFN-stimulated gene quantification
Supernatants collected 24 h after infection at a MOI of 3, and D0–D5 after infection at a MOI of 0.1, were used to quantify secreted cytokines. IFN-β, RANTES/CCL5, IP-10/CXCL10 and IL-1β were quantified using ALPHAlisa (Perkin Elmer), IFN-λ1 and thymic stromal lymphopoietin (TSLP) were quantified using standard ELISA (eBioScience). IFN-λ2/3, IFN-α, and IFN-γ were quantified by Milliplex assay (Millipore). RNA was extracted from cells infected at a MOI of 3, and RT-qPCR performed to quantify IRF7, Mx1 and IL-33 mRNA (see online supplementary information).
Statistical analysis
Normality of distribution of data was assessed with the Kolmogorov–Smirnov goodness-of-fit test. As data were not normally distributed, analyses were performed using generalised linear analyses on log-transformed data to determine cross-sectional associations between viral and host factors. Longitudinal analyses were performed for repeated measurements on each individual using mixed model. Correlation coefficients were calculated using the non-parametric Spearman rank test. All data analyses were conducted using STATA V.12 (Stata, College Station, Texas, USA).
Results
Wheeze and atopy were associated with reduced IFN production by NECs in response to RSV but not to hMPV
All cultures of NECs exposed to RSV at a MOI of 0.1 produced more IFN-β than their uninfected replicate cultures (figure 1A) (p<0.05). By D5, NECs from children with atopy (with or without wheeze) had secreted significantly less IFN-β compared to healthy cultures (p<0.05) (see online supplementary figure S1A). Overall, TECs produced less IFN-β than NECs in response to RSV regardless of subject group (figure 1B and see online supplementary figure S1B). However, reduced IFN-β expression was only detected on D3 in TECs from children with non-atopic wheeze compared to healthy TECs (p<0.05). Following infection with low MOI hMPV, NECs and TECs from children with wheeze/atopy generally produced similar or more, rather than less, IFN-β compared to healthy AECs by D5 (figure 1C,D), although these differences were not statistically significant (see online supplementary figure S1C,D).

Nasal epithelial cells (NECs) (A and C) and tracheal epithelial cells (TECs) (B and D) infected with 0.1 pfu/cell respiratory syncytial virus (RSV) (A and B), or 0.1 TCID50 units/cell human metapneumovirus (hMPV) (C and D), or uninfected. Secretion of interferon (IFN)-β was compared for infected (solid lines) and uninfected (dotted lines) replicate cell cultures. N values: NECs+RSV: healthy 4, wheeze alone 6, atopy alone 7, wheeze and atopy 6. TECs+RSV: healthy 5, wheeze alone 5, atopy alone 5, wheeze and atopy 7. NECs+hMPV: healthy 3, wheeze alone 5, atopy alone 7, wheeze and atopy 6. TECs+hMPV: healthy 5, wheeze alone 5, atopy alone 4, wheeze and atopy 7. Comparison between groups was analysed using STATA software and tests for non-parametric data; median±range, *p<0.05. #p<0.05 compared to uninfected control.
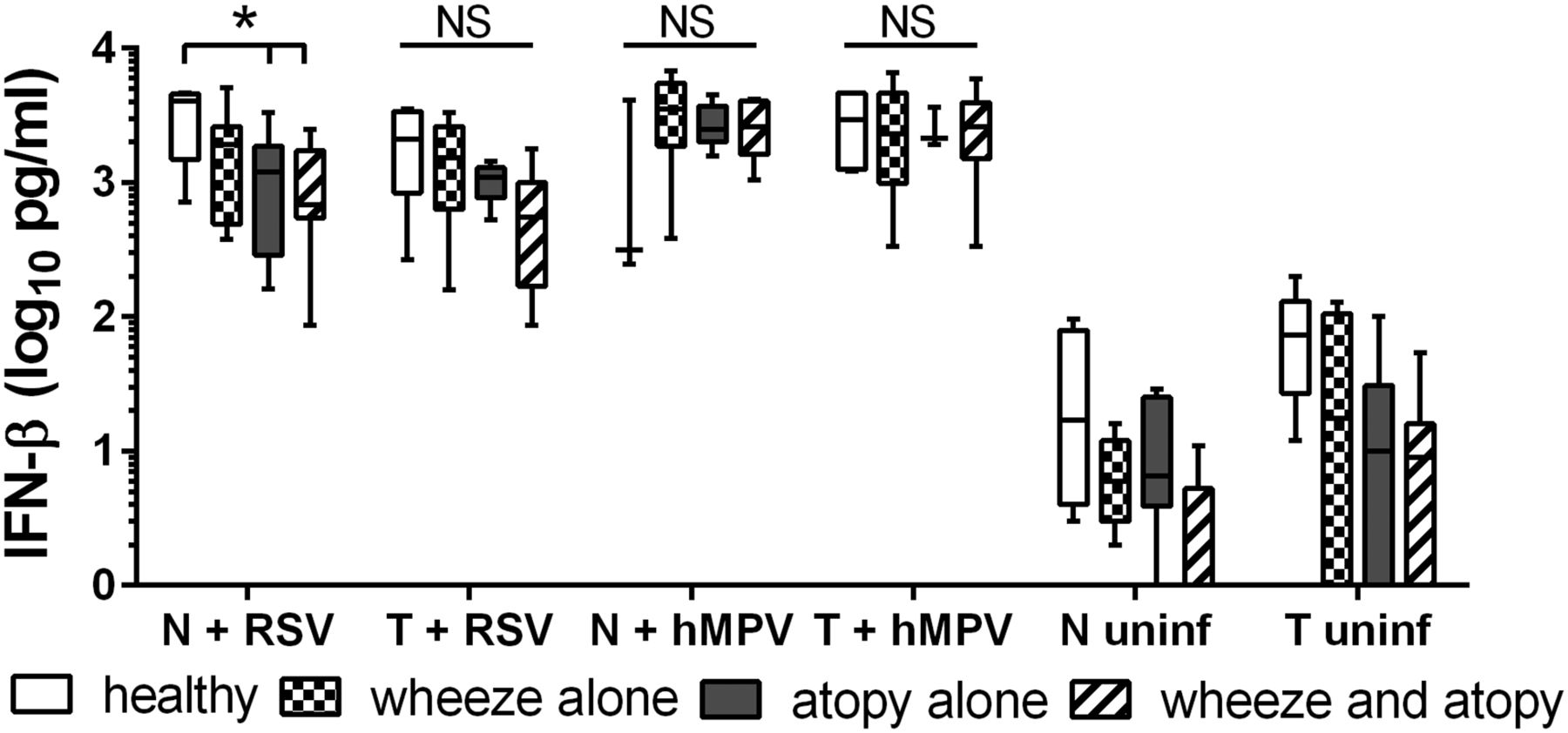
IFN-β was also reduced for NECs and TECs from children with wheezy or non-wheezy atopy compared to healthy NECs 24 h after RSV infection at a MOI of 3 (figure 2). However, this was statistically significant only for NECs (p<0.05). Following infection with hMPV at a high MOI, there was a trend for NECs from children with wheeze and/or atopy to produce more IFN-β than NECs from healthy children, while in TECs the induction of IFN-β was similar for all subject groups (figure 2).

Secreted interferon (IFN)-β and was quantified from nasal epithelial cells (NECs) and tracheal epithelial cells (TECs) 24 h postinfection at a high multiplicity of infection of 3 pfu/cell respiratory syncytial virus (RSV) or 3 TCID50 units/cell human metapneumovirus (hMPV), or uninfected. N values: NECs+RSV: healthy 4, wheeze alone 6, atopy alone 7, wheeze and atopy 6. TECs+RSV: healthy 5, wheeze alone 5, atopy alone 5, wheeze and atopy 7. NECs+hMPV: healthy 3, wheeze alone 5, atopy alone 7, wheeze and atopy 6. TECs+hMPV: healthy 5, wheeze alone 5, atopy alone 4, wheeze and atopy 7. Comparison between groups was analysed using STATA software and tests for non-parametric data. Bottom and top of box plots represent the 5th and 95th percentiles, bar represents the median, and whiskers represent minimums and maximums. *p<0.05.
There was a trend towards reduced IFN-λ1 secretion in response to RSV at low (figure 3A) and high (figure 3B) MOI for NECs and TECs, although this did not reach statistical significance. Conversely, there was a trend towards elevated IFN-λ1 associated with wheeze and atopy from NECs infected with low (figure 3A) and high (figure 3B) MOI hMPV, although this was not observed for TECs. There was no modulation of the IFN-λ2/3 response to RSV and hMPV based on disease status (see online supplementary figure S3A). IFN-α2 was not induced above baseline levels (see online supplementary figure S3B), and IFN-γ was not detected in any AEC culture.

Secreted interferon (IFN)-λ1 was quantified (A) 5 days postinfection from nasal epithelial cells (NECs) and tracheal epithelial cells (TECs) infected with 0.1 pfu/cell respiratory syncytial virus (RSV) or 0.1 TCID50 units/cell human metapneumovirus (hMPV), or uninfected, and (B) 24 h postinfection at a high multiplicity of infection (MOI) of 3 pfu/cell RSV or 3 TCID50 units/cell hMPV, or uninfected. N values: NECs+RSV: healthy 4, wheeze alone 6, atopy alone 7, wheeze and atopy 6. TECs+RSV: healthy 5, wheeze alone 5, atopy alone 5, wheeze and atopy 7. NECs+hMPV: healthy 3, wheeze alone 5, atopy alone 7, wheeze and atopy 6. TECs+hMPV: healthy 5, wheeze alone 5, atopy alone 4, wheeze and atopy 7. Comparison between groups was analysed using STATA software and tests for non-parametric data. Bottom and top of box plots represent the 5th and 95th percentiles.
Wheeze and atopy were associated with increased RSV and hMPV shedding, independent of the IFN response
RSV shed from NECs from children with wheeze and/or atopy increased steadily over 5 days following infection at a MOI of 0.1 to be approximately 5–10-fold higher than RSV shed from healthy NECs at D5 (figure 4A, see online supplementary figure S3A) (p<0.05). Despite reduced IFN-β secretion by these same cultures (figure 1A), correlation coefficient analyses demonstrated that there was no relationship between shed virus at D5 and IFN-β secretion at D5 (figure 5A) or earlier at D3 (figure 5B). RSV shed from TECs was not significantly different among subject groups (figure 4B, see online supplementary figure S3A), and was inversely correlated with secreted IFN-β at D5, although this correlation did not reach statistical significance (figure 5C). hMPV shed from NECs from atopic wheezy children reached a peak titre on D3 that was approximately 12-fold (1.2 log10) higher than healthy NECs (p<0.01). By D5, NECs from atopic and non-atopic wheezy children had shed more virus than NECs from healthy children (online supplementary figure S3B) (p<0.05, p<0.01). On D3, TECs from children with atopy (with or without wheeze) shed more hMPV than healthy TECs, although by D5 only TECs from children with wheeze and atopy shed more hMPV than healthy TECs (figure 4D, online supplementary figure S3B) (p<0.01). There was no correlation between IFN-β secretion and hMPV shed from NECs (figure 5D), however, there was a trend towards a positive correlation between IFN-β secretion and hMPV shed from TECs (figure 5E).
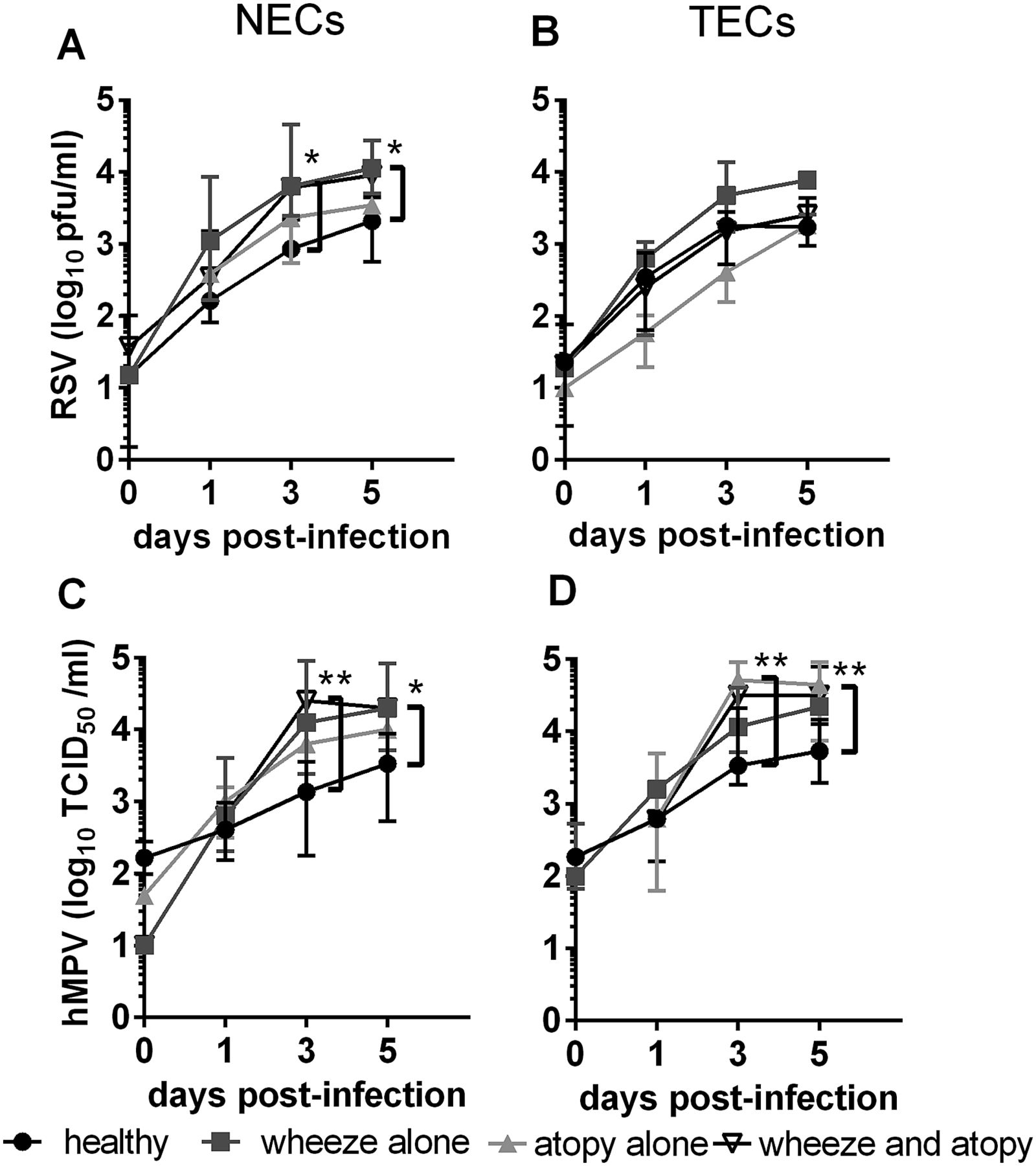
Virus shed into cell culture supernatants by nasal epithelial cells (NECs) (A and C) and tracheal epithelial cells (TECs) (B and D) infected with 0.1 pfu/cell respiratory syncytial virus (RSV) (A and B), or 0.1 TCID50 units/cell human metapneumovirus (hMPV) (C and D). N values: NECs+RSV: healthy 5, wheeze alone 5, atopy alone 7, wheeze and atopy 6. TECs+RSV: healthy 5, wheeze alone 5, atopy alone 5, wheeze and atopy 7. NECs+hMPV: healthy 4, wheeze alone 5, atopy alone 7, wheeze and atopy 5. TECs+hMPV: healthy 6, wheeze alone 5, atopy alone 4, wheeze and atopy 7. Comparison between groups was analysed using STATA software and tests for non-parametric data, median±range. *p<0.05, **p<0.01.

Correlation between secreted interferon (IFN)-β and shed virus following infection with 0.1 pfu/cell respiratory syncytial virus (RSV) or 0.1 TCID50 units/cell human metapneumovirus (hMPV). (A) Nasal epithelial cells (NECs)+RSV D5, (B) NECs+RSV IFN-β D3 and shed virus D5 (C) tracheal epithelial cells (TECs)+RSV, (D) NECs+hMPV, (E) TECs+hMPV. Spearman rank correlation.
Viral replication in AECs infected with RSV or hMPV at a high MOI followed a similar trend to shed virus after a low MOI, however, no statistical differences were detected among disease groups (see online supplementary figure S4).
Wheeze and atopy were not associated with modulated IFN signalling or inflammatory cytokine production in response to RSV or hMPV
Despite reduced IFN-β secretion in response to RSV infection (MOI 3) of NECs from children with atopy (with or without wheeze), this did not affect downstream expression of the ISGs, IRF7 and Mx1 (see online supplementary figure S5A). IRF7 was chosen as it is essential for the amplification of type I and III IFN and has been identified as an asthma ‘hub’ gene, while Mx1 was chosen as a representative canonical ISG with antiviral function. Interestingly, hMPV induced more IRF7 than RSV in TECs from all subject groups (p<0.05, p<0.01) and more IRF7 in TECs than NECs from children with atopic and non-atopic wheeze (p<0.05), suggesting virus and site-associated differences in response.
Wheeze and atopy were not associated with any difference in inflammatory cytokine production. The only cytokine that demonstrated a trend (although not statistically significant) to be reduced in association with wheeze/atopy was IL-1β in NECs infected with RSV (MOI 3; figure 6A). HMPV induced significantly more IL-1β in NECs and TECs than RSV, suggesting virus-associated differences in response. There was no difference in the expression of CXCL10, CCL5, or TSLP associated with disease status (figure 6B–D), nor was IL-33 mRNA elevated in response to viral infection (figure 6E).
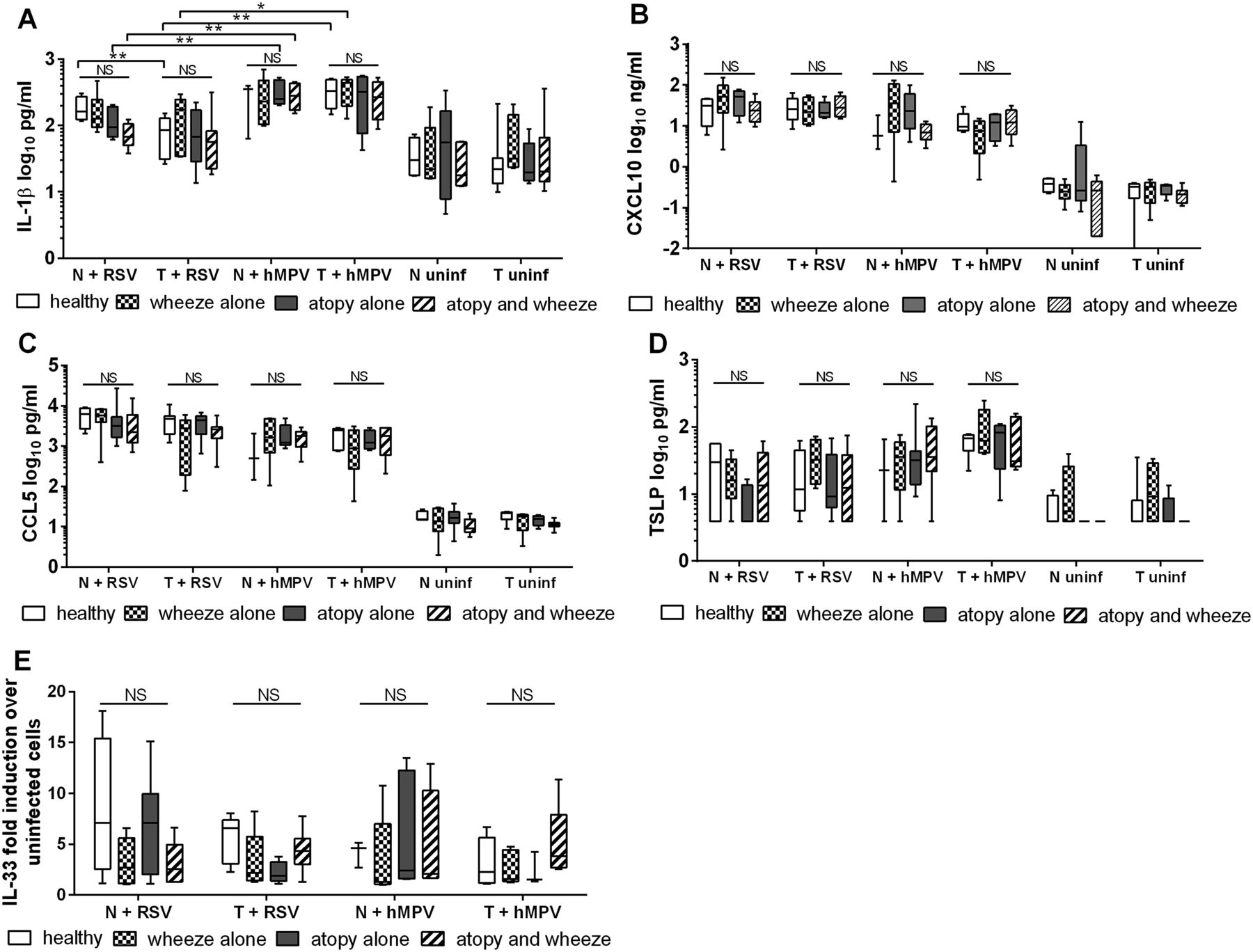
Secreted inflammatory mediators (A) IL-1β, (B) CXCL10, (C) CCL5, (D) TSLP were quantified from nasal epithelial cells (NECs) and tracheal epithelial cells (TECs) 24 h after infection with 3 pfu/cell respiratory syncytial virus (RSV) or 3 TCID50 units/cell human metapneumovirus (hMPV), or uninfected. (E) IL-33 gene expression was quantified by RT-qPCR of the same cells. N values: NECs+RSV: healthy 4, wheeze alone 6, atopy alone 7, wheeze and atopy 6. TECs+RSV: healthy 5, wheeze alone 5, atopy alone 5, wheeze and atopy 7. NECs+hMPV: healthy 3, wheeze alone 5, atopy alone 7, wheeze and atopy 6. TECs+hMPV: healthy 5, wheeze alone 5, atopy alone 4, wheeze and atopy 7. Comparison between groups was analysed using STATA software and tests for non-parametric data. Bottom and top of box plots represent the 5th and 95th percentiles, bar represents the median and whiskers represent minimums and maximums. *p<0.05, **p<0.01.
NECs and TECs responded similarly to viral infection regardless of disease status
We were interested in comparing the antiviral response of NECs and TECs regardless of disease status. When the time course of responses following infection at a MOI of 0.1 was plotted for each subject group, we found that IFN-β expression (see online supplementary figure S6A and B) and shed virus (see online supplementary figure S6C and D) were almost identical in TECs and NECs from all subject groups, except healthy individuals for which NECs produced more IFN-β than TECs. Due to unavailability of cells from both sites, or a lack of survival of matched cultures, only data from 13 RSV-infected and 10 hMPV-infected matched NEC and TEC cultures were available for correlation analysis. However, these data demonstrate that NECs and TECs from an individual respond similarly to viral infection regardless of disease status (figure 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Correlation between tracheal epithelial cells (TECs) and nasal epithelial cells (NECs) from the same individual regarding interferon (IFN)-β secreted on D5 following infection with (A) 0.1 pfu/cells respiratory syncytial virus (RSV) or (B) 0.1 TCID50 units/cell human metapneumovirus (hMPV). Spearman rank correlation. p<0.05 is considered significant.
Discussion
The results from the present study highlight the multifactorial nature of the antiviral response of AECs. The hMPV infection data presented here demonstrate that AECs from children at high risk of developing asthma, based on atopy and a history of early-life viral wheeze, are not intrinsically deficient in their ability to produce IFN in response to viral infection. It has been well published that AECs from atopic and non-atopic adults with asthma and children have a deficient IFN response to RV infection.9 ,10 ,13 However, our data suggest that this defect is not strongly evident in young, wheezy or atopic children in response to other viruses. This is similar to a recent study in which bronchial epithelial cells (BEC) from well-controlled atopic adults with asthma were not at all deficient in their ability to produce IFN in response to RV infection.16 ,17 It may be that an impaired IFN response of AECs is restricted to patients with severe asthma. Certainly, it has been shown that BECs from children with severe therapy-resistant asthma do not produce an effective antiviral response to RV infection.18 In the study presented here, we have infected paediatric AECs with RSV and hMPV, as both these viruses are common during early childhood and cause wheeze within the study cohort. We did not infect parallel cultures with RV, although this would be interesting to investigate, as there are no published data concerning experimental RV infections of AECs from very young pre-asthma-affected children.
The RSV data presented here, however, demonstrate virus-specific modulation of the IFN-β response associated with wheeze and atopy. RSV produces two non-structural proteins (NS1 and 2), which suppress type I and III IFN induction14 ,19 and signaling20–22 via several mechanisms. HMPV does not produce NS proteins, although other IFN antagonistic mechanisms have been reported.23–25 RSV and hMPV are detected principally by the cytosolic pattern recognition receptor RIG-I,26 ,27 which then interacts with the adaptor molecule IPS-1 to activate IFN-β transcription factors IRF3 and IRF7. Although RSV28 and hMPV24 proteins interfere with RIG-I–IPS-1 interactions, it may be that RSV is more effective at inhibiting this interaction such that an existing defect is augmented. However, as this phenotype was observed in RSV-infected NECs and not TECs, reduced IFN-β cannot be explained entirely by the action of NS1/2 proteins.
Elevated viral shedding associated with wheeze and atopy did not correlate to IFN-β secretion, and occurred regardless of a reduced (RSV) or elevated (hMPV) IFN-β response, particularly for NECs. This suggests an IFN-independent defect associated with wheeze and atopy. The lack of significant modulation of the IFN-λ response or downstream IFN signalling in AECs from children with wheeze/atopy also indicates an IFN-independent mechanism for increased viral replication and shedding. It is not clear from this study what this mechanism may be. There is evidence of delayed apoptosis in response to RV infection of BECs from patients with asthma,9 and a defect in the expression Rac1 in asthma-affected AECs leads to the inhibition of epithelial cell engulfment of apoptotic cells.29 ,30 AECs from patients with asthma also show an increase in the autophagy-related gene, ATG5, and increased autophagosome production.29 Perturbation of these pathways associated with wheeze and atopy requires further investigation, as it may be that intrinsic defects in programmed cell death pathways drive increased viral replication and shedding, with the IFN response being secondary and modulated by the specific infecting virus. Certainly, an increase in viral replication would result in an increase in RSV NS1/2 expression and, therefore, an increase in the IFN-β inhibitory effects of these proteins.
In this study, wheeze and atopy alone were associated with some key indicators of an impaired antiviral response, however, wheeze and atopy together had a synergistic effect on the antiviral response that resulted in increased viral shedding. This differs somewhat from a previous study in which a deficient immune response to RV infection was equally attributable to asthma and atopy, without additive effects of the two conditions.13
In the present study, we did not find differential regulation of TSLP, IL-33, or CCL5 associated with wheeze or atopy. TSLP and IL-33 are secreted by AECs to promote local Th2 inflammation associated with asthma.31 The lack of an association between asthma and regulation of inflammatory cytokine production has also been observed in BECs from adults with asthma in response to RV infection. However, this may be indicative of submerged cultures, as in one study of BECs from adults with asthma, in air-liquid interface culture secreted greater levels of TSLP than healthy BECs in response to RSV.32 Interestingly, we observed a trend towards reduced IL-1β secretion from NECs infected with RSV, which paralleled the reduction in IFN-β. This differs from published reports in which a reduction in IFN-β leads to an increase in IL-1β secretion.33 This may be indicative of RSV-specific modulation of the antiviral response. RSV is known to regulate apoptosis,34 and may also regulate IL-1β-driven cell death via pyroptosis. However, this requires further investigation.
Epithelial cells from the lower airways are difficult to source from very young children, and NECs have been proposed as a surrogate for AECs from the lower respiratory tract.35 We compared the IFN response of NECs and TECs within subject groups, and also from single individuals where possible. Although most other studies use lower airway cells of bronchial origin (BECs), it is unlikely that innate immune responses will differ critically between the trachea and major segmental bronchi. Although the numbers of NEC and TEC cultures available from the same individuals were small in this study, positive correlation for IFN-β production demonstrates that NECs can be used as surrogates for TECs regardless of disease status. Interestingly, however, on a population basis, we observed reduced IFN-β and increased viral shedding in response to RSV in NECs, but not TECs from children with wheeze and/or atopy. This suggests that intrinsic cellular defects, and RSV regulation of the IFN-β response may be more profound in the upper than the lower airway epithelium. RV12 and RSV36 replicated better in air-liquid interface cultures of healthy BECs than NECs, suggesting higher resistance to infection by upper than lower AECs. We also observed elevated IFN-β production by healthy NECs compared to healthy TECs, which may have highlighted the reduced IFN-β production of NECs from children with wheeze and atopy. As the nasal epithelium is exposed to viral infections before the lower respiratory epithelium, a defect in antiviral defence in this compartment may lead to increased susceptibility of the lower airways to RSV infection in children with wheeze and atopy.
In conclusion, data from the present study demonstrate that AECs, particularly NECs, from children with a history of wheeze and/or atopy have an intrinsic IFN-independent defect in the antiviral response that results in elevated viral replication. This in turn, exacerbates virus-specific regulation of the IFN response and may also explain why some viruses are more likely to be associated with asthma.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
KMS and EB contributed equally.
-
Contributors Conceived and designed the experiments: KMS, PDS and EF. Subject recruitment: CJ. Primary cell culture: JS, CS, FMP, EF and KMS. Performed experiments: KMS, EF and EB. Statistical analysis: LC. Manuscript preparation: KMS, EB, EF, SP and PDS.
-
Funding This work was supported by grants from the National Health and Medical Research Council, Australia (#1002035), the Office of Health and Medical Research, Government of Queensland (#50133) and Royal Children's Hospital Foundation, Brisbane, Queensland, Australia (#50005).
-
Competing interests None.
-
Ethics approval All aspects of the study were approved by the Institutional Review Boards of the Royal Children's Hospital and the University of Queensland.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement All data from the study are in the process of being submitted for publication.