Article Text

Abstract
Background Alpha-1-antitrypsin (A1AT) deficiency is the only recognised genetic risk factor for chronic obstructive pulmonary disease (COPD), a leading cause of morbidity and mortality worldwide. Since A1AT is the major inhibitor of neutrophil elastase (NE), this enzyme has become widely implicated in the pathogenesis of COPD in general; however, there is currently no specific biomarker for its pre-inhibition activity. Such a biomarker should be a measure of elastase-specific COPD disease activity with the potential to assess early targeted therapeutic intervention, in contrast to traditional and non-specific disease severity markers such as forced expiratory volume in 1 s.
Methods In pilot studies, plasma Aα-Val360 and markers of neutrophil activation were measured in 95 subjects with a range of A1AT concentrations. Aα-Val360 and sputum elastase activity were also measured in a further seven PiZ A1AT-deficient subjects over the course of an acute exacerbation. Finally, Aα-Val360 was measured in plasma from subjects randomised to receive A1AT replacement or placebo in the EXACTLE trial.
Results The plasma concentrations of Aα-Val360 and A1AT related exponentially, consistent with previous theoretical and in vitro experimental data. L-233 (an intracellular NE inhibitor) blocked generation of Aα-Val360 and subsequent A1AT/NE complex formation. Aα-Val360 was related to the spirometric severity of lung disease in A1AT deficiency, to sputum elastase activity in acute exacerbations and was decreased in subjects receiving A1AT replacement therapy (while remaining constant in those receiving placebo).
Conclusions Aα-Val360 represents the first specific footprint of pre-inhibition NE activity and is a potential biomarker of disease activity and progression in subjects with elastase-dependent COPD.
Trial registration The EXACTLE study was registered in ClinicalTrials.gov as ‘Antitrypsin (AAT) to Treat Emphysema in AAT-Deficient Patients’; ClinicalTrials.gov Identifier: NCT00263887.
- Alpha 1-antitrypsin
- pulmonary disease
- chronic obstructive
- leukocyte elastase
- biological markers
- alpha1 antitrypsin deficiency
- COPD mechanisms
- COPD exacerbations
- emphysema
- neutrophil biology
Statistics from Altmetric.com
- Alpha 1-antitrypsin
- pulmonary disease
- chronic obstructive
- leukocyte elastase
- biological markers
- alpha1 antitrypsin deficiency
- COPD mechanisms
- COPD exacerbations
- emphysema
- neutrophil biology
Key messages
What is the key question?
Is a specific measure of pre-inhibition neutrophil elastase activity a valid biomarker of COPD disease activity?
What is the bottom line?
Aα-Val360 is a neutrophil elastase specific biomarker that relates to COPD disease activity and treatment and may fulfil the essential criteria of a biomarker.
Why read on?
Biomarkers relevant to the pathological processes of COPD are urgently needed.
Introduction
Chronic obstructive pulmonary disease (COPD) is a slowly progressive group of conditions for which there is no specific treatment. The pathophysiology is relatively poorly understood, although it is widely accepted that inflammation is a central process that results in tissue destruction.1 Emphysema is a feature of many patients with COPD and is characteristic of patients with the genetic deficiency of alpha-1-antitrypsin (A1AT). This protein is the major inhibitor of neutrophil elastase (NE), which has led to the general acceptance that emphysema in these subjects is a direct consequence of failure to control elastase released by neutrophils as they migrate into the lung.2 This is supported by the observation that NE causes many of the features of COPD in animal models,3 especially emphysema.4
The development of emphysema centralises on the destruction of lung elastin.5 However, although there is some evidence that this occurs in man,3 detection of free NE is not usually possible (in the absence of acute inflammation and only then in airway secretions) as the enzyme is rapidly inactivated by its cognate inhibitors, especially A1AT. The destructive process therefore occurs in a confined microenvironment at the site of neutrophil degranulation and has been termed ‘quantum proteolysis’. There is a high concentration of NE in the azurophil granules and therefore physiological concentrations of inhibitor cannot inactivate the enzyme at the point of release.6 As the enzyme diffuses away, its concentration falls exponentially and activity is quenched at molar equivalence by A1AT. This exponential relationship explains both the obligate area of proteolytic destruction during neutrophil activation and a greatly enhanced one as A1AT levels fall below 11 μmol (as in A1AT deficiency). This is also consistent with the particular susceptibility of homozygous Z A1AT-deficient subjects to developing emphysema (while those with an SZ phenotype who have an average A1AT concentration of 11 μmol or more are probably not7), and possibly the severity of ANCA-positive neutrophil-mediated tissue damage in renal vasculitis and necrotising panniculitis (both associated with A1AT deficiency8 9).
The evaluation of the extracellular activity of elastase in both physiological and pathological states has been hampered by the difficulty in developing a specific assay of the in vivo activity of this rapidly inhibited enzyme. Detection of NE using immunoassays identifies and quantifies both free enzyme and enzyme bound to its inhibitors. However, this provides no direct evidence of the destructive potential of NE at the point of degranulation. Alternative assay procedures have therefore been pursued which depend upon the detection of a footprint of NE activity by measuring NE cleavage products, including urinary desmosine (a major cross-linking amino acid in elastin) and elastase-generated elastin peptides. However, neither is specific to NE and may even be influenced by diet,10 renal function11 and reflect tissue breakdown in many organs12 which may (at least partly) explain the lack of association between urinary desmosine and forced expiratory volume in 1 s (FEV1)13–15 and the failure to decrease in PiZ individuals receiving A1AT augmentation.16 17 Such drawbacks can be overcome by assays of NE-specific cleavage products of other natural macromolecular substrates.
The flaws of current markers of COPD disease severity are increasingly recognised and there is an urgent need for new disease markers. For example, the day-to-day variation of FEV1 within individuals may be greater than its decline over several years,18 and pharmacological interventions may significantly increase FEV1 yet fail to influence mortality.19 Also, FEV1 and other current markers of COPD disease severity are measures of end-organ damage. In contrast, a biomarker of disease activity should not only relate (at least in part) to cross-sectional disease severity, but also predict future progression and offer the potential for proof of concept and dose ranging intervention prior to the development of irreversible lung disease.
This paper describes a novel assay based on NE-specific cleavage of fibrinogen that is related to neutrophil degranulation from both normal subjects and those with A1AT deficiency. This unique assay provides a footprint of NE activity in the microenvironment of activated neutrophils and may thus provide an ideal biomarker for the activity of NE-mediated diseases.
Methods
The Aα-Val360 assay
NE cleaves human fibrinogen at multiple sites (US patent No. 6124107, see figure 1 in online supplement). We chose the Aα-Val360 site to develop an assay because this larger fibrinogen fragment is likely to be more stable than relatively small peptides due to the disulfide network of the protein. The full methodology for the Aα-Val360 assay is given in the online supplement.
Study subjects
Subjects gave informed consent and were studied in the stable clinical state at least 8 weeks after any acute infection. The project was approved by the local research ethics committee (LREC 3359).
For the pilot study (15 PiZ A1AT-deficient subjects, 12 normal subjects), blood was drawn into two lithium heparin tubes and 150 μM calcium ionophore A23187 (Sigma, St Louis, Missouri, USA) was added to one. After 60 min at 37°C, both tubes were centrifuged (500 g × 10 min) and the plasma collected. All samples were processed within 90 min of collection and stored at −70°C until analysed.
For the second part of the study (40 PiZ A1AT-deficient subjects, 24 normal subjects, 4 with partially-deficient SZ phenotype), untreated plasma alone was processed.
In a further study, subjects with PiZ A1AT deficiency and chronic bronchitis were assessed during an acute exacerbation as reported previously.20 Sufficient plasma samples remained in seven subjects to assess Aα-Val360. Sputum elastase activity was measured using the synthetic substrate methoxysuccinyl-ala-ala-pro-val-paranitroanilide (MeOSAAPVpNa; Sigma Aldrich, Poole, UK) (see online supplement).21
The EXACTLE trial was a double-blind randomised controlled trial of A1AT replacement versus placebo in patients with PiZ A1AT deficiency. All gave informed consent and the study was approved by the ethics committee of the Capital Region of Denmark (Hillerød, Denmark), the National Health Service West Midlands South Birmingham research ethics committee (Birmingham, UK) and the regional ethics committee of Lund (Lund, Sweden). No subject had received A1AT augmentation prior to the trial. Plasma samples were obtained at the baseline visit and after 6 months of treatment, and frozen at −70°C until analysed. The previously reported primary outcome measure for the EXACTLE trial was CT densitometry.22
Measurement of A1AT/NE complex, myeloperoxidase and calprotectin
Full details are given in the online supplement.
Statistical analysis
Full details are given in the online supplement.
Results
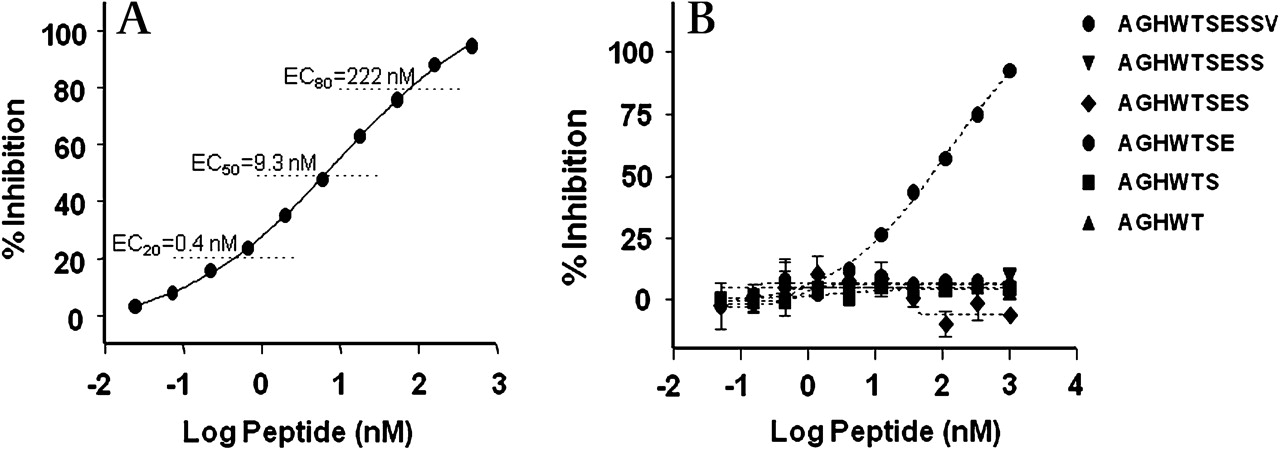
The europium-based ELISA for Aα-Val360 possessed a dynamic range of approximately 550-fold (figure 1A). Figure 1B shows that any synthetic C-terminal truncated peptide lacking the C-terminal Val residue was not recognised by the antisera. To determine if fibrinogen fragments that might be generated by cleavage distal to the Val360 terminal site would be recognised by the antisera, a peptide spanning the Val360-Ser361 elastase cleavage site was prepared. The data in Figure 2A in the online supplement show a more than 300-fold loss in recognition by the antisera. The within-and-between plate assay coefficients of variation were 9.6% and 23.6%, respectively.

Specificity of Aα-Val360 antisera. (A) Antibody inhibition curve by the peptide sequence of the neo-epitope. (B) Loss of inhibition by truncated epitopes even with the single terminal valine deleted.
In order to determine the selectivity of NE to produce the Aα-Val360 neoepitope, a series of proteinases were analysed for their ability to hydrolyse fibrinogen at the Aα-Val360-Ser361 site. Elastase incubated at an enzyme to fibrinogen ratio of 1:250 hydrolysed fibrinogen to form Aα-Val360 neoepitope in a time-dependent manner (see Figure 3A in the online supplement). The human serine proteinases plasmin, trypsin, cathepsins B, H and L did not generate the Aα-Val360 neoepitope. Proteinase 3 (PR3) is a serine proteinase with 54% sequence homology to elastase co-localised in the neutrophil azurophil granule. The incubation of fibrinogen with PR3 resulted in the time-dependent formation of Aα-Val360 at approximately 15% of the elastase catalysed rate, suggesting a potential but slight contribution of PR3 to Aα-Val360 in vivo.
Production of Aα-Val360 and A1AT/NE complex in human blood with A23187 and inhibition with a cell penetrant elastase inhibitor
Figures 3A and 3B in the online supplement show the concentration-dependent and time-dependent production of Aα-Val360 and A1AT/NE complex formation in human blood stimulated with the calcium ionophore A23187. The maximal amount of Aα-Val360 and A1AT/NE complex formation was produced within 30–60 min at a final concentration of 75 μM A23187. This concentration of A23187 did not cause the release of lactate dehydrogenase, suggesting that elastase was released by exocytosis from azurophilic granules of neutrophils into the plasma rather than cell death. Other stimuli such as zymosan and phorbol myristate acetate gave similar results (data not shown). These findings demonstrate that the cleavage of an endogenous substrate by NE released from neutrophils occurs in blood, even in the presence of high concentrations of the natural inhibitors A1AT and α2-macroglobulin.
Figure 2A shows the structure of a mechanism-based elastase inhibitor Merck L-233 (patent WO/2009/045419; Merck, Rahway, USA). Figure 2B shows that L-233 blocks both A1AT/NE complex formation and Aα-Val360 production in A23187-stimulated blood with an EC50 of0.2 μM and 0.4 μM, respectively, which is equivalent to the concentration of elastase in blood. An important distinguishing feature between L-233 and A1AT is the ability of L-233 to penetrate neutrophils and inhibit elastase intracellularly (figure 2C). In this experiment whole blood was incubated with 3H-L-233, followed by isolation of neutrophils. These data confirm the ability of L-233 to bind NE intracellularly and also demonstrate that subsequent stimulation does not release bioactive NE generating Aα-Val360 or A1AT/NE complex.
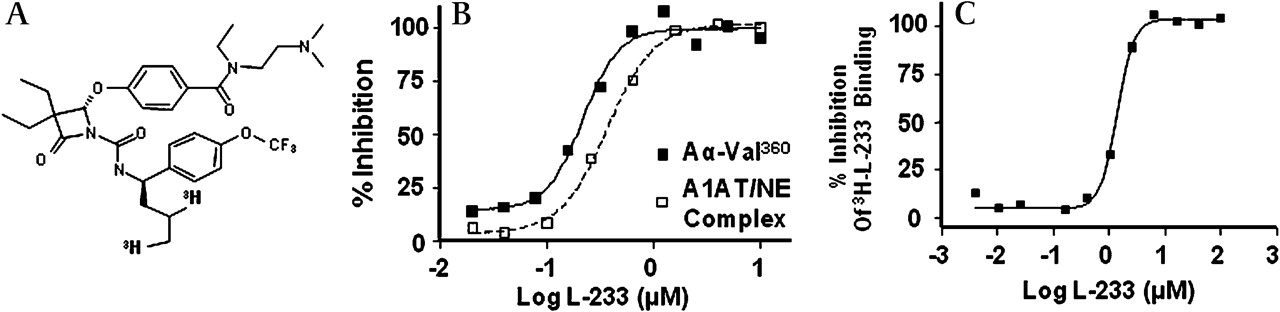
L-233, a cell penetrant inhibitor of neutrophil elastase (NE), completely blocks Aα-Val360 formation. (A) Structure of L-233. (B) Complete inactivation of NE (prior to its release) occurs in the presence of 1 μM L-233, as demonstrated by the complete abrogation of the formation of alpha-1-antitrypsin/neutrophil elastase (A1AT/NE) complexes and Aα-Val360. Human whole blood was preincubated with increasing concentrations of L-233 at 37°C for 60 min, A23187 was then added, followed by a further incubation period of 60 min. Plasma was prepared and Aα-Val360 and A1AT/NE complex measured by immunoassay. (C) Further evidence of the cell penetrant properties of L-233. Human whole blood, with 3H-L-233 and increasing concentrations of unlabelled L-233, was incubated at 37°C for 60 min. The red blood cells were removed by lysis with hypertonic saline, the leucocytes were pelleted by centrifugation and bound drug was determined in a scintillation counter.
Identification of the Aα-Val360 in healthy control and PiZ plasma samples
Aα-Val360 was detectable in all samples with values from 3.5 to 18.9 nM. The concentration showed no relationship to circulating neutrophil numbers in either those with normal A1AT (r=0.115; p=0.736) or PiZ A1AT deficiency (r=0.066; p=0.688). However, in the pilot study, stimulation of the neutrophils by the calcium ionophore A23187 after venepuncture led to a marked increase in Aα-Val360 associated with a similar increase in plasma myeloperoxidase and calprotectin concentrations (table 1). These data support the concept that the Aα-Val360 signal reflects (at least partly) baseline neutrophil activation.
Mean (SE) results from the pilot study for plasma Aα-Val360, myeloperoxidase (MPO) and calprotectin (CP) for subjects with normal alpha-1-antitrypsin (A1AT) and those with A1AT deficiency of the PiZ phenotype
In the second part of the study involving more subject samples, Aα-Val360 showed a positive correlation with both myeloperoxidase and calprotectin in unstimulated samples from both healthy controls and A1AT-deficient subjects (table 2). Importantly, the Aα-Val360 concentration in these two groups showed a positive correlation with the A1AT/NE complex (table 2 and figure 3), confirming that the cleavage product reflects the release of active NE before it is inhibited by A1AT.
Spearman correlation coefficients (r) and significance (p) for Aα-Val360 with alpha-1-antitrypsin/neutrophil elastase (A1AT/NE) complex, myeloperoxidase (MPO) and calprotectin (CP) for subjects with and without A1AT deficiency
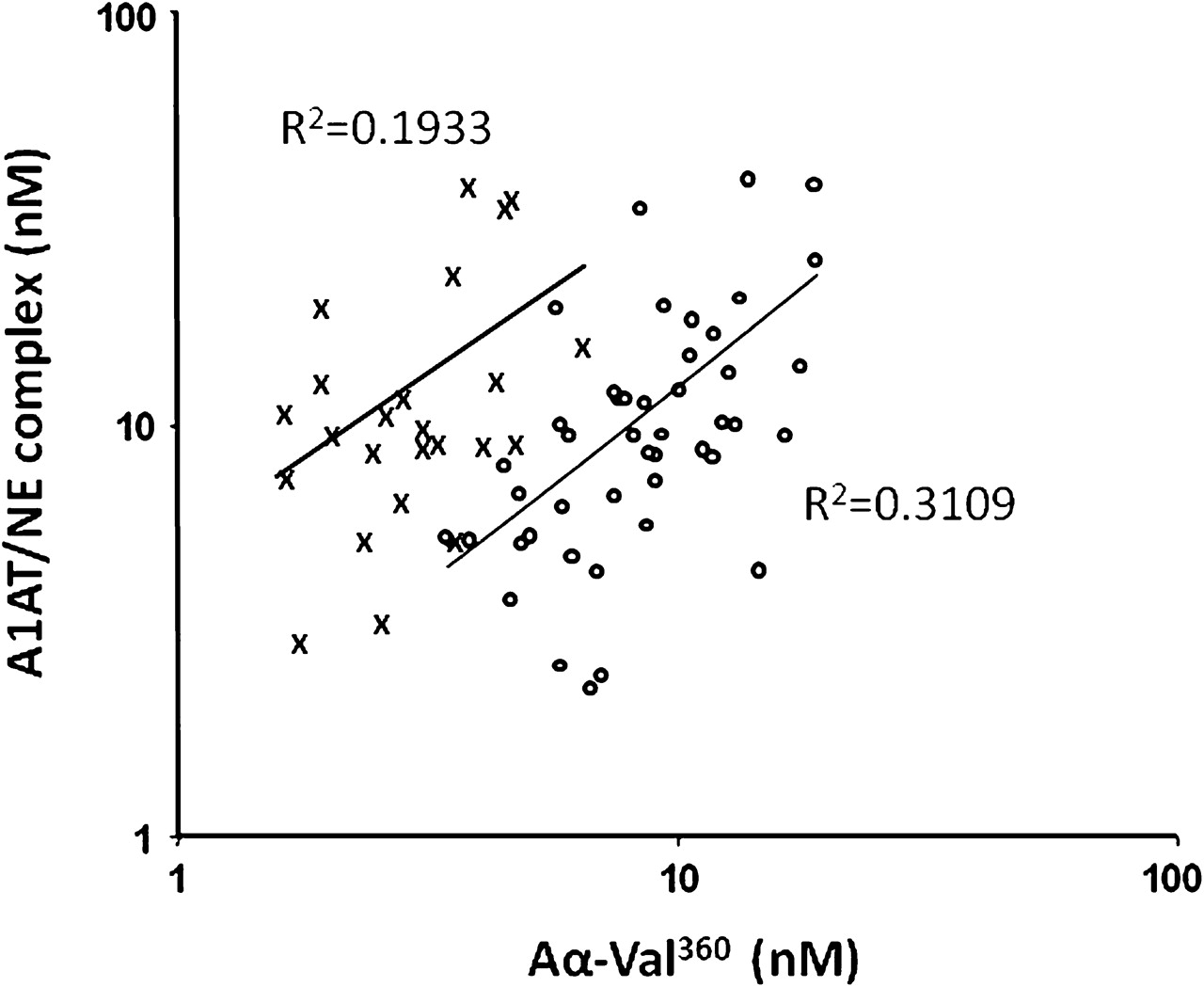
Positive correlations of alpha-1-antitrypsin/neutrophil elastase (A1AT/NE) complexes and Aα-Val360 in subjects with and without PiZ A1AT deficiency.The relationship between Aα-Val360 and the A1AT/NE complex is shown for subjects with (open circles) and without (crosses) A1AT deficiency. The correlation coefficients (r) are shown and the regression lines included. Although the relationship is similar in both groups (p<0.001 and p<0.025, respectively), the shift of the A1AT-deficient curve to the right indicates more generation of Aα-Val360 before NE is inhibited in subjects with A1AT deficiency.
In addition, the Aα-Val360 concentrations were markedly increased in the plasma of A1AT-deficient subjects (n=40; p=2.76×10−14) with a mean (SE) of 9.28 (0.61) nM compared with a mean (SE) of 3.16 (0.25) nM in those with normal A1AT (n=24), despite the similar amount of NE released (mean concentrations of A1AT/NE complex 11.57 (1.40) μM and 13.05 (1.96) μM for A1AT-deficient subjects and those with normal A1AT, respectively; p=0.487). In support of this biological effect of A1AT deficiency, the mean myeloperoxidase concentration showed a slight but marginal increase (21.12 (1.94) nM and 15.62 (2.05) nM, respectively; p=0.042).
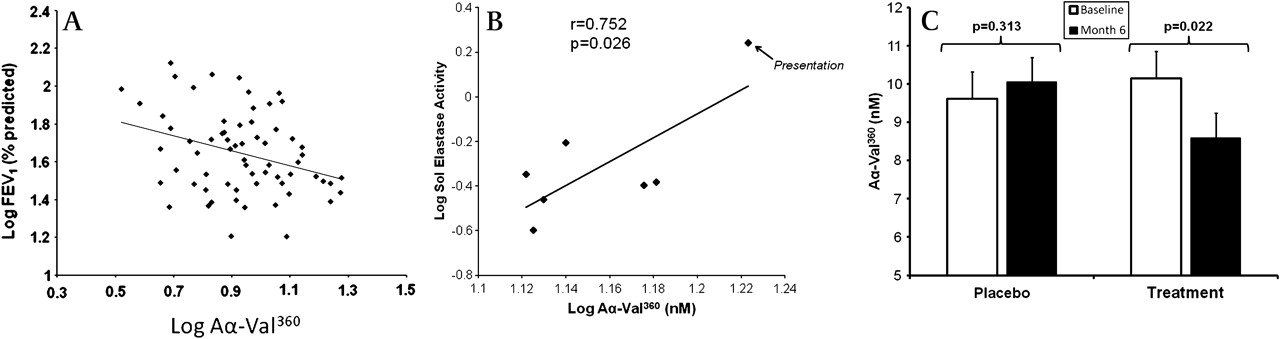
Importantly, the relationship between the Aα-Val360 concentration and the A1AT/NE complex concentration (a measure of NE release) was different for subjects with normal A1AT than for those with PiZ severe deficiency (figure 3). Indeed, overall the Aα-Val360 concentration showed an exponential relationship with the plasma A1AT concentration (figure 4A), with a marked increase in samples with a plasma A1AT concentration <11 μM. The Aα-Val360 level also showed a negative relationship with the FEV1 percentage predicted (figure 5A), with higher levels in those with worse lung function.
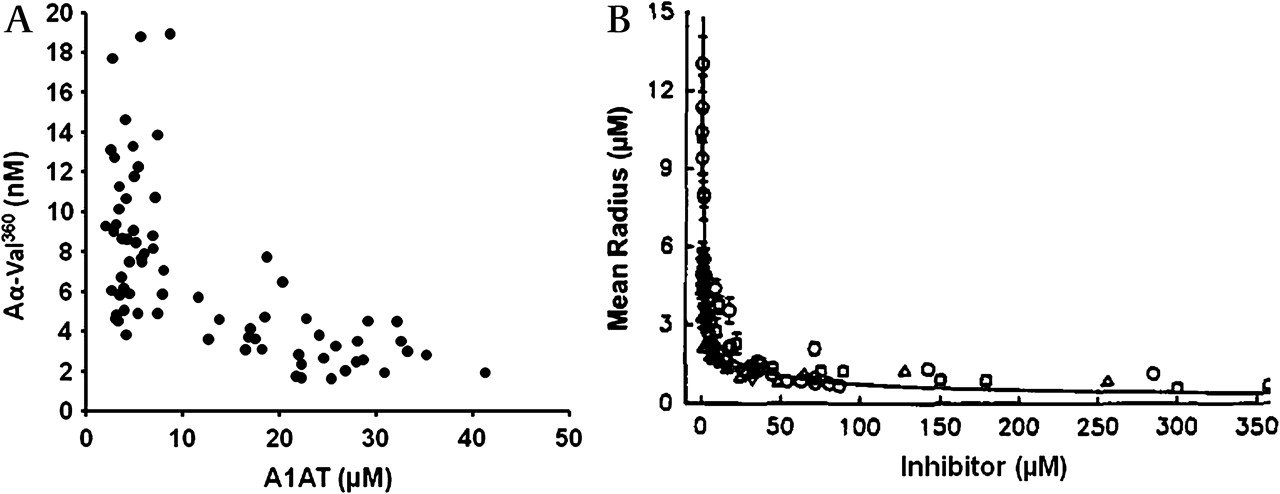
Quantum proteolysis. (A) Relationship between Aα-Val360 (nM) and plasma alpha-1-antitrypsin (A1AT) concentration in μM. (B) Comparison with in vitro data of the area of proteolytic degradation by neutrophils in the presence of increasing concentrations of neutrophil elastase (NE) inhibitors. Reprinted with permission from Liou and Campbell.23 © 1995, American Chemical Society.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Aα-Val360 relates to lung function and sol elastase activity and is decreased in PiZ subjects receiving alpha-1-antitrypsin (A1AT) replacement therapy but not in the placebo group. (A) Relationship between Aα-Val360 and forced expiratory volume in 1 s (FEV1) (% predicted) is shown on a double log scale. The correlation line is drawn (r=−0.321; p=0.005). (B) In this further study, the plasma Aα-Val360 concentration and sputum elastase activity were measured in seven PiZ A1AT-deficient subjects with chronic bronchitis during an acute exacerbation. Each point on the figure represents the mean Aα-Val360 concentration and elastase activity on an individual day. Both parameters were highest at presentation and fell progressively until the final day of the study (at which point the patients were clinically stable). The coefficient and significance is shown. (C) Aα-Val360 was measured in plasma obtained from 77 PiZ A1AT-deficient participants in the EXACTLE trial (a double-blind randomised controlled trial of placebo (n=34) versus A1AT replacement therapy (n=33)). The mean Aα-Val360 (with SE bar lines) is shown for subjects before the start of the trial (open histogram) and at 6 months (solid histogram) while receiving placebo or A1AT replacement therapy. The significance of the reduction in the Aα-Val360 on the active compound is shown. The Aα-Val360 was also higher in subjects in the placebo group than in those in the treatment group at month 6, although this did not quite achieve statistical significance (p=0.0835).
In a further study of PiZ-deficient subjects there was a high plasma Aα-Val360 concentration at the onset of an exacerbation which decreased over 28 days. Similarly, sputum elastase activity was greatest at the start of the exacerbation and fell as the episode resolved.20 The average results of Aα-Val360 for each of the collection days was related to the sputum sol phase elastase activity (figure 5B).
Analysis of plasma samples from a randomised controlled trial of A1AT augmentation (EXACTLE22) showed no difference between the two groups at baseline. There was a reduction in Aα-Val360 in the treatment group (p=0.022; 95% CI for difference −3.05 to −0.07) but not in the placebo group (p=0.313; 95% CI for difference −1.36 to 2.23) at 6 months (figure 5C). The mean Aα-Val360 was also higher in subjects in the placebo group than those in the treatment group at 6 months, although this just fell short of statistical significance (p=0.084). The Levene test showed that there was no significant difference in the variance from the mean between month 0 and month 6 in the placebo (p=0.500) or treatment group (p=0.670).
Discussion
We describe a unique assay based on the generation of an NE-specific cleavage product of fibrinogen which demonstrates good within and between assay variability. The Aα-Val360 correlated well with two other independent markers of neutrophil activation, especially following stimulation—namely, myeloperoxidase which is stored and released from the same granule as NE and a cytoplasmic protein calprotectin. In vitro activation of neutrophils in whole blood resulted in an increase in the plasma concentrations of all three signals. However, Aα-Val360 is a specific marker of pre-inhibition elastase activity and, although it is generally related to other neutrophil activation markers and total elastase release as measured by the A1AT/NE complex, it also provides unique information. The current study shows that the relationship between the pre-inhibition NE activity (as quantified by Aα-Val360) and the total amount of NE released (A1AT/NE complex) is different in subjects with A1AT deficiency than in those without, confirming the more specific nature of this marker.
Importantly, Aα-Val360 must be generated at the point of release of NE (before NE is captured and inhibited by A1AT), confirming the mathematical predictions23 and in vitro observations6 described by others. This reflects the exceptionally high concentrations of NE at the point of release from the granule that exceeds the concentration of surrounding A1AT. Quantum proteolysis, demonstrated by mathematical and in vitro modelling, predicts this effect would be increasingly marked as the concentration of A1AT falls below 11 μM, producing an exponential relationship between activity and radius. The in vivo data presented here (figure 4) confirm that the theory and in vitro data are correct in predicting the phenomenon.
The implications therefore are that, during neutrophil activation and migration, NE release produces an area of obligate proteolysis that leads to tissue damage thought to be central to the pathophysiology of emphysema. Furthermore, the enhanced proteolytic activity in subjects with an A1AT level <11 μM explains the more severe, early onset and rapid decline in lung function seen in subjects with A1AT deficiency. The evidence that this relates to in vivo degranulation is confirmed by the use of a specific intracellular inhibitor L-233 which completely abrogates the generation of Aα-Val360 and A1AT/NE complex by the neutrophil activator A23187 in a time- and concentration-dependent manner.
Aα-Val360 therefore supports the mathematical and in vitro data of Campbell et al6 23 and is a surrogate marker of activity central to neutrophilic COPD disease processes. However, to be a valid biomarker, Aα-Val360 must also24:
relate to disease severity;
be stable, but vary with events associated with disease progression;
be sensitive to effective intervention factors;
predict progression;
improve upon existing disease markers.
We showed that Aα-Val360 relates cross-sectionally to FEV1 and, while the limitations of FEV1 as a severity marker of COPD are recognised,24 25 this physiological parameter nevertheless relates well to mortality.26 Although this correlation is consistent with Aα-Val360 reflecting the disease processes that influence FEV1, it should be noted that interpretation is more complex. A high level of an NE marker could reflect the current status (severity), the process that results in the current status or future disease progression. Although FEV1 is a poor surrogate of pathological processes such as emphysema, a single cross-sectional value represents both disease activity and duration. Thus, further studies are needed to explore this relationship, which should include the relationship to other parameters of the emphysematous process (gas transfer and lung densitometry). In addition, the relationship of the marker to future progression needs to be assessed in a large group of patients over several years.
It is possible that a synergistic relationship exists between Aα-Val360 and fibrinogen since fibrinogen may also relate to COPD disease severity.27 However, although NE is released at a high concentration which exceeds that of most substrates, the total amount released is low and therefore the abundant fibrinogen protein remains in excess. Fibrinogen is therefore only likely to influence Aα-Val360 formation if the clotting factor concentration varies between individuals by several orders of magnitude. However, published data have shown that this is not the case, either between individuals in the stable state27 or within individuals experiencing an acute exacerbation.28 Furthermore, stimulation of neutrophils in plasma with ionophore greatly increases Aα-Val360, indicating the fibrinogen concentration is not a limiting factor (at least in the stable state).
We also demonstrated an increase in Aα-Val360 during exacerbations with a related rise in sputum elastase activity. These episodes are associated with faster disease progression,29 fulfilling the second criterion for a biomarker, and therefore would also be consistent with an NE-related process driving progression. However, in the absence of an acute exacerbation, sputum NE activity is either absent, below or close to the lower limit of detection21 and always absent in plasma which precludes direct use of NE activity as a biomarker in the stable state, which again supports the need for an alternative pre-inhibition marker of activity such as Aα-Val360.
The Aα-Val360 concentration decreased specifically in the A1AT-deficient subjects after 6 months of augmentation therapy but remained unchanged in those in the placebo group, thus demonstrating stability and an effect of treatment. The baseline Aα-Val360 values were identical in both groups and, although not powered to assess the specific effect of therapy on Aα-Val360, the results showed a trend to lower values in the treated group at 6 months, providing data to power future studies using this marker as an outcome. The efficacy of augmentation therapy has yet to be proven beyond doubt, although observational data30 and the effects on progression of CT densitometry are consistent with efficacy.22 31 32 The reduction in Aα-Val360 may therefore provide a short-term surrogate of efficacy of antielastase therapy in this disease and hence has the potential for dose ranging studies, although further validation of clinical outcomes will clearly be required.
These in vivo associations therefore provide background and circumstantial evidence required to validate Aα-Val360 as a biomarker of the NE-related pathophysiological process (at least in COPD related to A1AT deficiency). Since it is now widely accepted that the neutrophil and protease–antiprotease imbalance also play a role in the pathophysiology of COPD unrelated to A1AT deficiency, it is highly likely that Aα-Val360 will also be a valid biomarker in usual COPD. However, further cross-sectional and longitudinal studies are required to confirm this and to determine whether baseline levels of Aα-Val360 predict subsequent progression and/or relate better to clinical outcomes other than FEV1 in subjects with COPD (both with and without A1AT deficiency).
Further clinical data are required from ongoing studies to consolidate these initial findings, nevertheless Aα-Val360 represents the first specific footprint and marker of pre-inhibition elastase activity and therefore NE-dependent disease activity, rather than a measure of disease severity assessed at a single time point. It is worth speculating that Aα-Val360 could also be elevated in other diseases where increased neutrophil activation is implicated such as cystic fibrosis, acute myeloid leukaemia, idiopathic myelofibrosis and the ANCA-positive vasculitides. Clearly, prospective studies of carefully characterised patients will be necessary to explore these possibilities.
References
Supplementary materials
Web Only Data thx.2010.154690
Files in this Data Supplement:
Footnotes
Funding Talecris Biotherapeutics provided an unrestricted grant to the ADAPT (Antitrypsin Deficiency Assessment and Programme for Treatment) project to support this and ongoing research into alpha-1-antitrypsin deficiency.
Competing interests A patent covering L-233 (WO/2009/045419) is held by Merck, Rahway, NJ, USA. PEF and RAM are named inventors on this patent. A patent covering Aα-Val360 (US patent No 6124107) is held by Merck, Rahway, NJ, USA. PD, JLH and RAM are named inventors on this patent. Talecris Biotherapeutics has provided unrestricted grants to the ADAPT project for ongoing research into alpha-1-antitrypsin deficiency and sponsored the EXACTLE trial but had no involvement in the production of this manuscript.
Ethics approval Ethical approval for studies relating to patients on the ADAPT (Antitrypsin Assessment and Programme for Treatment) Registry was provided by the University Hospitals Birmingham NHS Trust research ethics committee. The EXACTLE trial was a double blind randomised controlled trial of alpha-1-antitrypsin replacement therapy versus placebo in patients with PiZ alpha-1-antitrypsin deficiency. All patients gave informed consent and the study was approved by the ethics committee of the Capital Region of Denmark (Hillerød, Denmark), the National Health Service West Midlands South Birmingham research ethics committee (Birmingham, UK) and the regional ethics committee of Lund (Lund, Sweden).
Provenance and peer review Not commissioned; externally peer reviewed.