Article Text

Statistics from Altmetric.com
Biomarkers need to fulfil several distinct requirements before they can be considered a valid indicator of chronic diseases such as COPD
Chronic obstructive pulmonary disease (COPD) has become recognised as a priority area for management of healthcare resources and development of new therapeutic strategies. This is based largely on economic burden and the excessive morbidity and mortality associated with the condition. The result has been a profusion of publications in recent years, many of which start with the observations that “COPD is currently the fifth and by the year 2020 will become the third or fourth leading cause of morbidity and mortality worldwide”.
More recently, the COPD literature has entered a second phase. This has arisen from the appreciation that COPD is more than a respiratory inflammatory condition and is associated with manifestations outside the lung. This has led to the concept that COPD is a systemic disease and has resulted in a rapid increase in papers exploring this aspect. Initial studies were primarily based on the association of reduced body mass index with severe COPD and common inflammatory pathways have been implicated.1 In particular, the central role of tumour necrosis factor (TNF)α has been proposed,2 and muscle biopsies in patients with COPD have shown apoptotic changes within skeletal muscle3 thought to be the result of the systemic inflammation.
In addition to the association of skeletal muscle dysfunction, it is also being appreciated that other co-morbidities such as cardiovascular disease,4 type II diabetes5 and osteoporosis6 are more commonly associated with patients with COPD than the general population. Indeed, the inflammatory basis for these other conditions is also gradually becoming appreciated, and there are many common pathogenic processes between them and COPD.1
Research in COPD is now entering its third phase. Many pharmaceutical companies are becoming involved in drug discovery programmes based on the development of new treatments to modulate the inflammatory processes in COPD. However, as with all chronic diseases, the progression of COPD is slow but continuous. Thus, not only does the complexity of the inflammatory pathway present a challenge to research workers and pharmaceutical companies, but also the conventional pathway of progressing from drug discovery through phase I and phase II to phase III controlled clinical trials is impaired by the lack of early “read outs” in phase II.
The traditional surrogate for progression in COPD is a physiological measurement (forced expiratory volume in 1 s (FEV1)) which can vary day-to-day more than the overall progression over several years. Thus, although many drugs have been developed and marketed based on FEV1 and evidence of symptomatic relief, the progression of the disease has remained unaltered. As interventions related to progression require many years of physiological follow-up, pharmaceutical companies have been hampered by the lack of specific or surrogate markers (closely linked to the pathogenic process in COPD) that are sensitive to facilitate short-term phase II proof of concept studies. Since these are the key to subsequent investment in large and lengthy phase III studies, there is an urgent need to identify such biomarkers. Understanding the inflammatory process involved in the pathophysiology of chronic diseases such as COPD provides the potential to identify more robust surrogate markers of the disease process that are sensitive to short-term interventions.
Nevertheless, identifying relevant biomarkers—although the key—is only the first step and thereafter validation becomes critical. Despite this, there is now a profusion of papers looking at both the local and systemic inflammatory processes in COPD, often compared with healthy smokers, patients with COPD who are undergoing exacerbations of their disease (when inflammation increases) and comparing these markers with lung function. This has, however, raised a significant problem of how to interpret such studies.
Although many potential mechanisms have been implicated in the pathophysiology of COPD, the role of proteolytic enzymes has been dominant for over 40 years. This is related to the initial observation that patients with α1-antitrypsin deficiency (a genetic deficiency of a key serine proteinase inhibitor) were particularly susceptible to developing emphysema. Since animal models showed that the instillation of enzymes normally controlled by α1-antitrypsin into the lungs produced emphysema, the proteinase/antiproteinase theory of COPD has evolved.7 This is based on the concept that proteolytic enzymes can produce many of the clinical and pathological features of patients with COPD, and that inflammatory processes lead to the release of proteolytic enzymes within the lungs. Excess release of such protein-ases, or a net reduction in the inhibitors required to control them, would lead to persistent enzyme activity and tissue damage producing the features of COPD.7
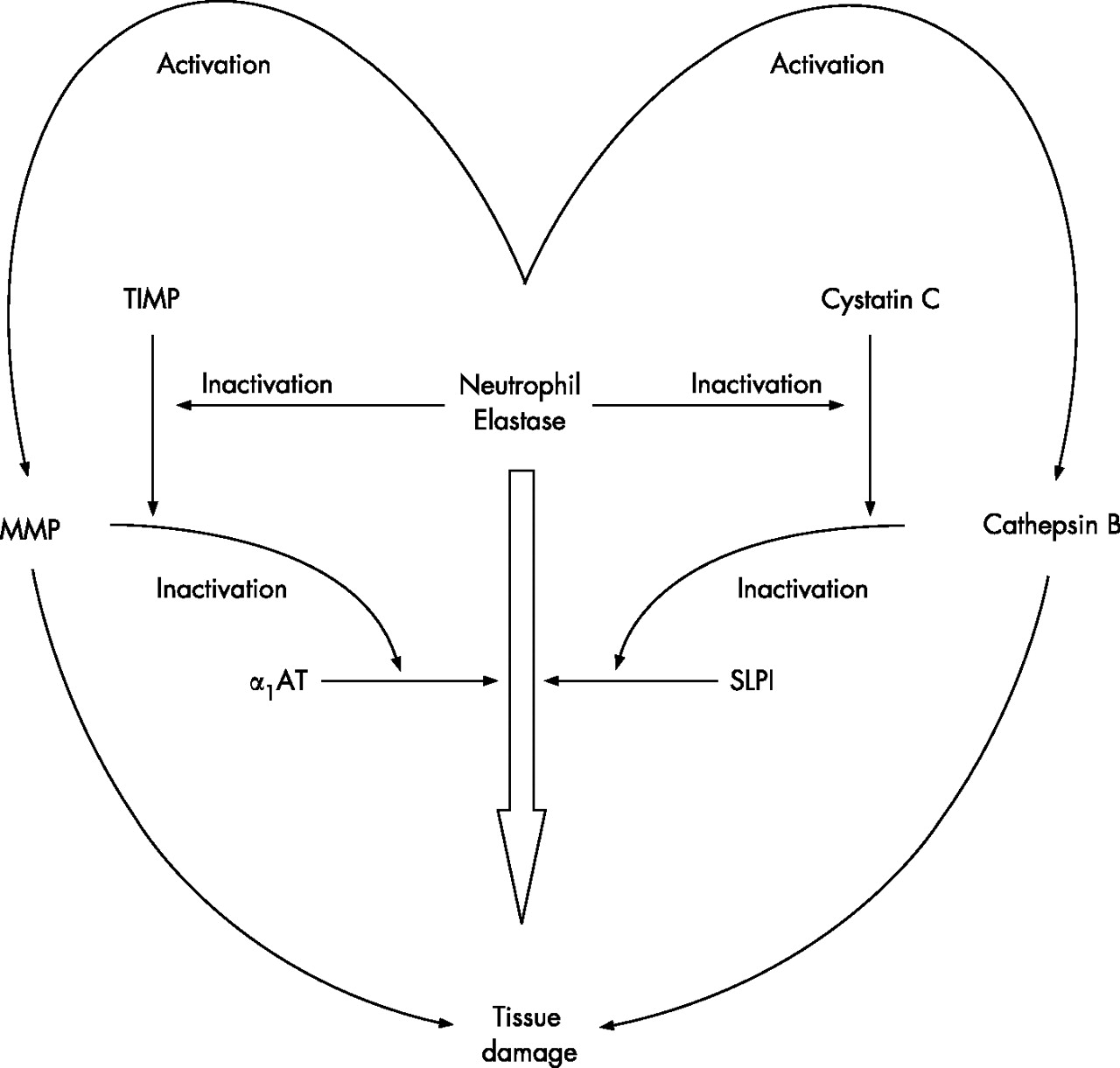
This simple concept initially related to serine proteinases and the serine proteinase inhibitors α1-antitrypsin (produced by the liver) and secretory leucoprotein-ase inhibitor which is produced locally. However, it has since become clear that such a straight pathway is an oversimplification and a cascade of proteinases and proteinase inhibitors interact, even if the common final pathway is tissue damage caused by neutrophil serine proteinases (fig 1). Furthermore, the use of a series of transgenic or knock-out mice has indicated that this protease cascade is linked to other critical steps in the inflammatory pathway, including TNFα and its receptor.8 For this reason, TNFα itself may be an important biomarker for the link between inflammation and tissue damage, and TNFα has been implicated in the systemic effects of muscle wasting.9 However, taking this forward provides a significant challenge of relating a local problem within the lung to a systemic inflammatory process where sampling and co-morbidities create confounding issues. In addition, the validation of biomarkers suffers from the problems of cross-sectional versus longitudinal observations, and whether the markers being identified are predictive or reflective of the disease process being studied.
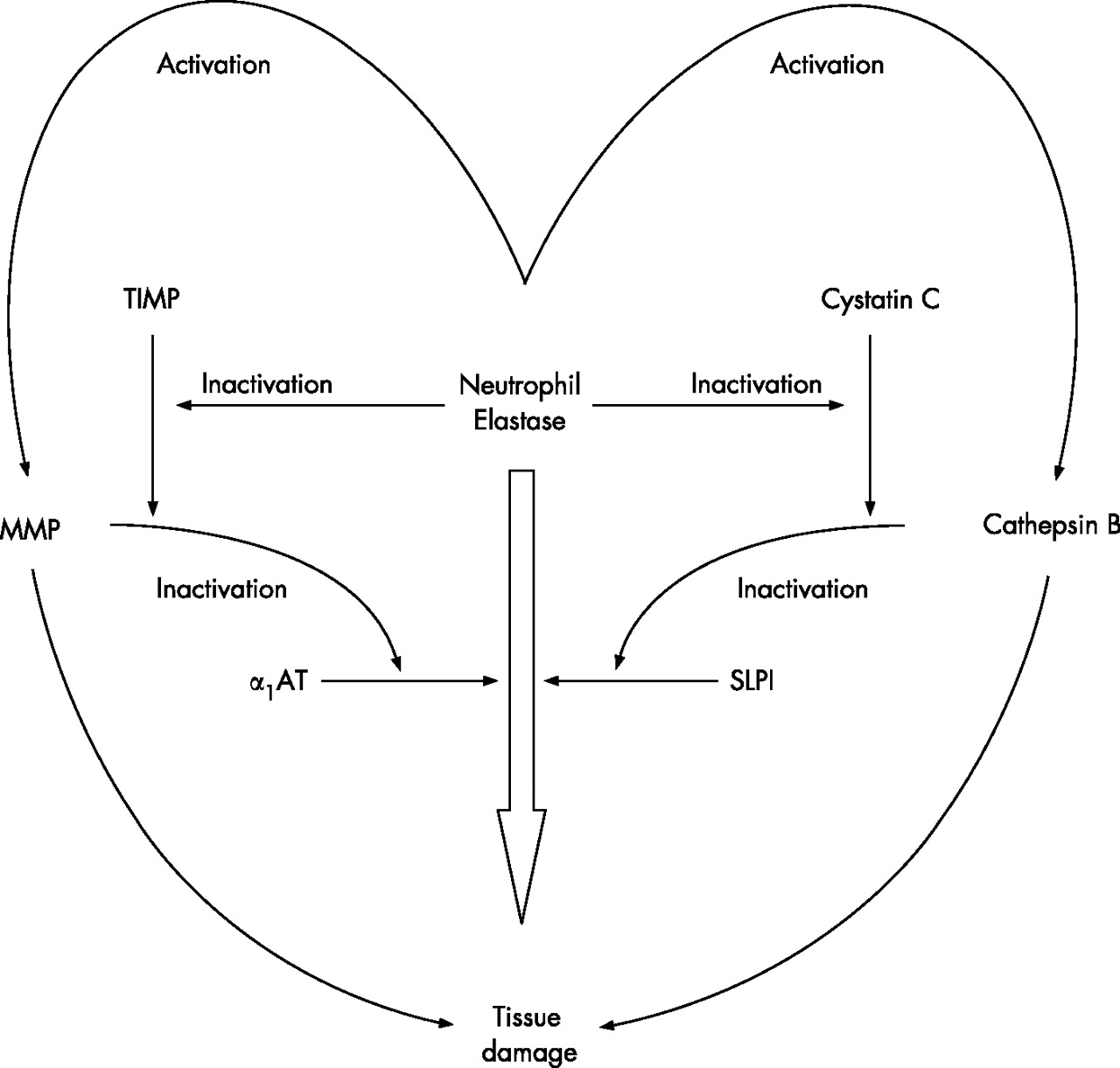
Proteinase and proteinase inhibitor cascade showing the inter-relationship between cysteine, serine and metalloproteinases. Individual proteinases can activate proteinases in another class and, at the same time, inactivate the relevant inhibitors. The net result is a cascade where the presence of several classes of proteinase can facilitate the end result of damage by an individual enzyme or in an additive way. MMP, matrix metalloproteinase; α1-AT, α1-antitrypsin; TIMP, tissue inhibitor of metalloproteinases; SLPI, secretory leucoproteinase inhibitor.
An ideal biomarker is a direct indicator of the process being studied. It has to be reproducible, easily measured and would have to be sensitive to effective interventions. For instance, TNFα has been implicated in the pathogenic processes of insulin resistance in humans.1 This concept is supported by studies of the obese insulin resistant mouse which showed that antagonism of TNFα leads to the development of insulin sensitivity.10 TNFα has also been implicated in osteoporosis,1 and specific interventions not only lead to an increase in bone thickness but also a reduction in TNFα production.11 In vascular disease, several recent studies have indicated that measuring serum C-reactive protein (CRP) predicts future vascular events,12,13 and CRP has also been implicated in the pathophysiological pathway leading to atheroma.1 Studies have also shown that statins not only reduce cholesterol but can reduce CRP,14 which could account for the subsequent reduction in cardiovascular events.
With this as a background, similar approaches are now being undertaken in COPD. For instance, CRP in the plasma is increased in COPD15 and can predict the likelihood of future outcomes,16 although it is difficult to implicate this acute phase protein in the pathophysiology of COPD. Indeed, it is more likely to be secondary as it is known that interleukin (IL)-6 stimulates CRP production in hepatocytes and IL-6 is increased in COPD.17 However, although IL-6 is a pro-inflammatory cytokine, it also has yet to be clearly implicated in the pathophysiological processes in COPD. The dysjunction of this approach has been highlighted recently in a study demonstrating that CRP has predicted mortality in COPD.16 The accompanying editorial clearly reminds readers that this may not reflect mortality from COPD (and hence pathophysiology of the disease) but, rather, other vascular events.18
There have been many studies of numerous other mediators in COPD which relate them to lung function, although often studies apparently showing a correlation almost certainly reflect two populations (ie, healthy smokers with one range for the mediator and patients with COPD who have reduced FEV1 and a different range). However, some studies do show a relationship between increasing concentrations of mediators and decreasing lung function.19,20 Nevertheless, even with such relationships, the concern regarding whether this is cause or effect remains a key issue.
Lung function in COPD shows progression with time but does not follow a straight line. As the FEV1 is a measure of airflow within the airways, there has to be a major change in obstruction of the small airways before the FEV1 is affected. Thus, lung damage in COPD can progress for a long time before the FEV1 declines, and many subjects with a normal FEV1 are not at risk.
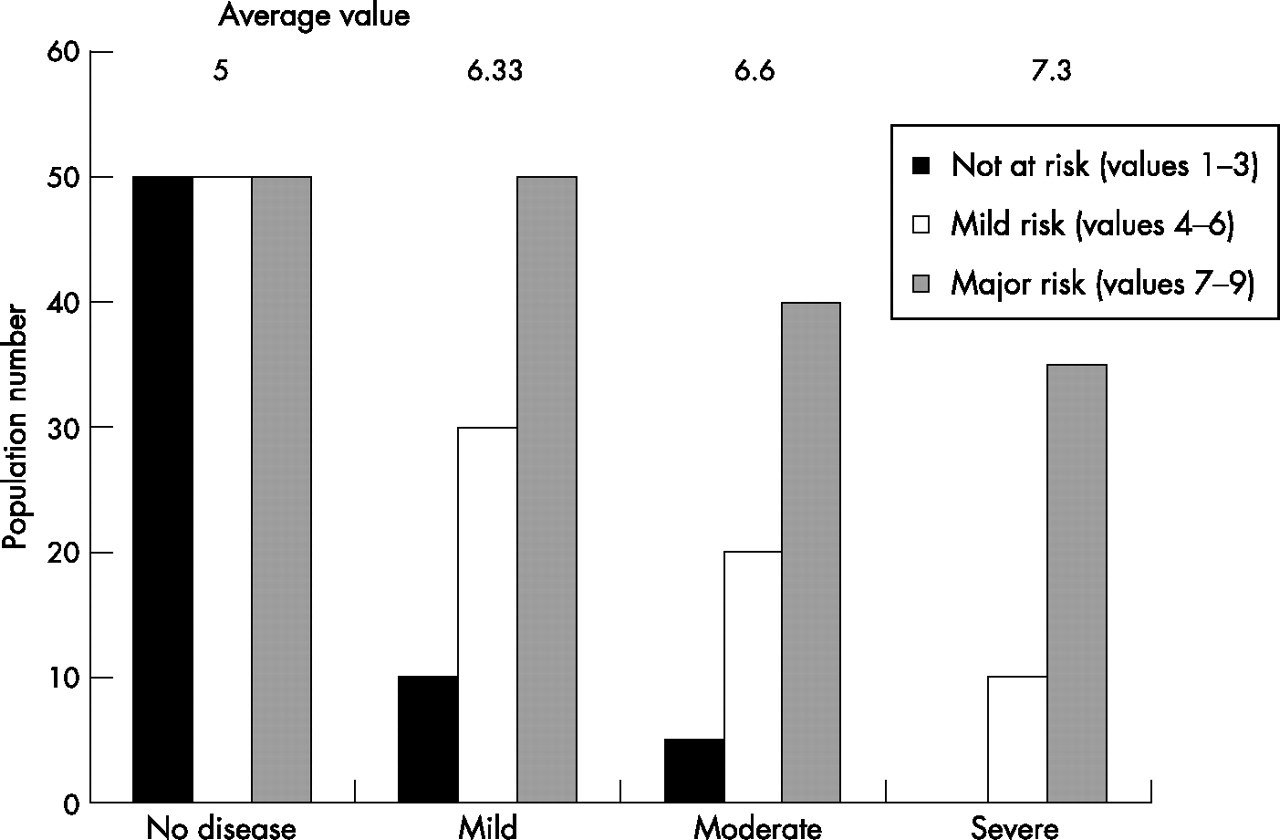
Once the FEV1 starts to decline, it does so rapidly in subjects at risk. However, in the presence of severe disease, rapid decliners are less likely to survive and long-term follow-up only relates to patients whose FEV1 decline has stabilised (the survivor effect). This produces the characteristic sigmoid curve for decline in FEV1, and this observation has raised questions about the nature of the relationship of biomarkers to lung function. The argument has been that any biomarker that shows an increased value related to FEV1 in a cross-sectional study is more likely to be reflective than predictive. However, even in cross-sectional studies, that is not necessarily the case. Figure 2 shows how an individual biomarker of susceptibility to disease progression relates to the severity of disease in cross-sectional studies. If individuals with chronic disease have varying degrees of low grade inflammation, the biomarker may be largely within the normal range as for cardiovascular disease and CRP. Before the disease becomes physiologically detectable, measurements would represent three different populations: those not particularly at risk, those with a moderate risk and those who are specifically at risk. As varying degrees of severity of the disease are studied, those most at risk would become relatively more over-represented in the cohort. This would result in an increase in the average concentration of the biomarker being studied towards the level of those most at risk. Thus, such data may provide initial confidence that the biomarker being studied is related to the disease process.
{kind=link}
{kind=link}
Diagrammatic representation of the relationship between the average biomarker concentration and severity of lung disease. The data are related to three populations of individuals at no risk with low levels of the biomarker, those at intermediate risk with intermediate levels and those at high risk with high levels. In cross-sectional studies, as the disease severity increases the proportion of patients in the high-risk group also increases, leading to a rise in the average biomarker level for the group.
However, further studies are then clearly necessary to provide final reassurance. This requires longitudinal studies relating the initial biomarker to subsequent progression, as has been shown with airway mediators in COPD,20 and is likely to be easier if the mediator plays a recognised role in the pathophysiology rather than a general role in inflammation. With this concept in mind, it is difficult to understand or reconcile two recent studies showing that baseline CRP relates to the decline in FEV1 in mild to moderate COPD21 but not in population screening.22 Perhaps, unlike cardiovascular disease, biomarkers in lung disease are only informative once the disease is established. Clearly these recent studies emphasise the complexity of biomarker studies during this period of their infancy.
The study of biomarkers is of major importance in developing new therapeutic strategies for chronic slowly progressive diseases. However, to be valid, the biomarker needs to fulfil several distinct requirements.
-
It must be central to the pathophysiological process implicated in the disease.
-
It must therefore reflect or be a very clear surrogate of that disease process.
-
It must be stable and only vary with events known to relate to disease progression.
-
Those at risk with a higher value at baseline must become more prevalent as disease severity or its prevalence increases.
-
The biomarker must predict progression.
-
The biomarker must also be sensitive to intervention factors that are known to be effective; this may prove the biggest hurdle.
However, once all these boxes are ticked, the biomarker can be used as an early read out of therapeutic efficacy with a reasonable degree of confidence.
In summary, before we get carried away with continued measurements of a multitude of factors that have some link to inflammation in COPD, it is important that we take a deep breath and rethink our concepts of the inflammatory process and design and deliver our studies appropriately and with care.
Biomarkers need to fulfil several distinct requirements before they can be considered a valid indicator of chronic diseases such as COPD
REFERENCES
Footnotes
-
Competing interests: None.