Article Text

Abstract
Purpose To assess efficacy and safety of a new dry powder formulation of inhaled colistimethate sodium in patients with cystic fibrosis (CF) aged ≥6 years with chronic Pseudomonas aeruginosa lung infection.
Study design and methods A prospective, centrally randomised, phase III, open-label study in patients with stable CF aged ≥6 years with chronic P aeruginosa lung infection. Patients were randomised to Colobreathe dry powder for inhalation (CDPI, one capsule containing colistimethate sodium 1 662 500 IU, twice daily) or three 28-day cycles with twice-daily 300 mg/5 ml tobramycin inhaler solution (TIS). Study duration was 24 weeks.
Results 380 patients were randomised. After logarithmic transformation of data due to a non-normal distribution, adjusted mean difference between treatment groups (CDPI vs TIS) in change in forced expiratory volume in 1 s (FEV1% predicted) at week 24 was −0.98% (95% CI −2.74% to 0.86%) in the intention-to-treat population (n=373) and −0.56% (95% CI −2.71% to 1.70%) in the per protocol population (n=261). The proportion of colistin-resistant isolates in both groups was ≤1.1%. The number of adverse events was similar in both groups. Significantly more patients receiving CDPI rated their device as ‘very easy or easy to use’ (90.7% vs 53.9% respectively; p<0.001).
Conclusion CDPI demonstrated efficacy by virtue of non-inferiority to TIS in lung function after 24 weeks of treatment. There was no emergence of resistance of P aeruginosa to colistin. Overall, CDPI was well tolerated.
Trial Reg No EudraCT 2004-003675-36.
- Cystic Fibrosis
- Bacterial Infection
- Inhaler devices
- Nebuliser therapy
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Key messages
What is the key question?
-
The key question is whether colistimethate formulated as a dry powder inhaler can be as effective as inhaled antibiotics given via a nebuliser in controlling chronic P aeruginosa infection in cystic fibrosis patients.
What is the bottom line?
-
In our study we established that Colobreathe® dry powder inhaler (CDPI) was as effective as tobramycin nebuliser solution for inhalation (TNSI).
Why read on?
-
In addition to efficacy we also investigated patient response to the delivery devices and changes in microbiological sensitivity over time: the results may be important to cystic fibrosis patients.
Introduction
Patients with cystic fibrosis (CF) are highly susceptible to infection with Pseudomonas aeruginosa, with chronic pulmonary infection occurring in over 80% of adult patients.1 ,2 Chronic P aeruginosa infection accelerates decline in lung function3 ,4 and effective management of P aeruginosa is therefore necessary to maintain lung function for as long as possible.5 ,6
Inhaled colistimethate sodium with oral ciprofloxacin is effective in the treatment of P aeruginosa,7 limiting deterioration of lung function in patients with chronic infection.8–11 Current treatment guidelines recommend colistimethate sodium as one treatment option12 but administration via older nebulisers is lengthy6 and complex, and may have a negative effect on adherence.13 ,14
Colobreathe dry powder for inhalation (CDPI) uses a new encapsulated dry-powder formulation of micronised colistimethate sodium administered via a convenient hand-held inhaler (Turbospin, PH&T, Milan, Italy) (figure 1). The aim was to increase patient convenience, which may improve treatment adherence and clinical outcomes.15 CDPI uses capsules containing the equivalent of 125 mg colistimethate sodium (1 662 500 IU) in fine particle form, with no excipients. Average deposition of one inhaled capsule is at least the average deposition of a nebulised dose (not less than 2 MU).16 The inhaler has low internal resistance compatible with the flow rate typically achieved by this patient group and drug deposition is maintained over a range of flow rates.16 ,17

Turbospin® inhaler and appearance of Colobreathe packaging.
The device is portable, and may increase the proportion of drug delivered to the lung compared with nebulisation. It requires little cleaning, no maintenance, and is disposable at the end of each 28-day pack.
The Freedom study was set up to investigate the efficacy and safety of this new dry powder formulation of inhaled colistimethate sodium (CDPI) compared with tobramycin nebuliser solution for inhalation (TIS) in patients with CF aged ≥6 years with chronic P aeruginosa lung infection.
Methods
The study was carried out in 66 CF centres in Europe from March 2003 to October 2007. Approval was sought from Competent Authorities and Independent Ethics Committees. The study was carried out according to the requirements of the Declaration of Helsinki 1964 with subsequent amendments and the International Conference on Harmonisation/Good Clinical Practice. Written informed consent or assent was obtained from all patients, or from carers of children. The trial registration number is EudraCT 2004-003675-36.
Patients
The study enrolled patients with CF aged ≥6 years, with forced expiratory volume in 1s (FEV1) of 25–75% predicted,18 chronic P aeruginosa lung infection and a stable clinical condition. The definition of chronic P aeruginosa infection was for at least 50% of sputum samples (minimum of three) to be positive for P aeruginosa over the 12 months prior to the first day of trial medication or for two positive samples over the 6 months prior to the first day of trial medication. Stable clinical condition was defined as there being no evidence of a current acute respiratory exacerbation at the pre-run visit, and in the investigator's opinion, no likelihood of an acute respiratory exacerbation at visit 1 (start of treatment).
The main exclusion criteria were the presence of Burkholderia cepacia complex infection in the airways, ongoing pulmonary exacerbation (based on a modified Fuchs definition) and sensitivity to any study medication.19
Study design
This was a prospective, centrally randomised, phase III, open-label study. The primary endpoint was assessed by independent assessors of pulmonary function unaware of individual patient therapy.
Before randomisation, all patients had to undergo at least two 28-day TIS on–off cycles, as previous treatment or as preparation for the study. Patients were then randomised equally either to continuous treatment over a 24-week period with CDPI (one capsule of 1.6625 MU twice daily), or to three 28-day courses of TIS (300 mg/5 ml tobramycin, twice daily, Novartis Pharmaceuticals, Basel) using a PARI LC Plus nebuliser with a suitable compressor. Each course of TIS was followed by a 28-day off period.
Normal patient standards of care were maintained but additional ad hoc anti-pseudomonal agents were generally not permitted, apart from management of acute respiratory exacerbations for long-term prophylaxis with chronic oral agents such as ciprofloxacin, or as planned courses of elective oral or intravenous therapy.
Short-acting inhaled bronchodilators were not permitted within the 2 h prior to pulmonary function testing.
Study assessments
The main efficacy measure was the change in mean FEV1% predicted from baseline at week 24. The Knudson correction was used for all pulmonary function assessments.18
Other efficacy measures included the following:
-
Susceptibility of respiratory tract P aeruginosa isolates from both groups to colistin and tobramycin. Testing was carried out at a single central laboratory using the eTest (AB Biodisk (now bioMérieux SA, Marcy l'Etoile, France)). Minimum inhibitory concentration (MIC) breakpoints (based on British Society for Antimicrobial Chemotherapy recommendations 2001) were used to determine antimicrobial sensitivity.20 For colistin these were ≤4 mg/litre susceptible, >4 mg/litre resistant, and for tobramycin, ≤2 mg/litre susceptible, ≥8 mg/litre resistant. MIC50 and MIC90 values were derived (MIC of antibiotic needed to inhibit 50% and 90% of a given culture respectively).
-
Change in forced vital capacity (FVC, litre), change in FEV1 (litre/s), change in forced expiratory flow between 25% and 75% of the vital capacity (FEF25–75, litre/s).
-
Compliance with study medication, assessed by counting used and unused medications at study end.
The incidence and intensity of all adverse events (AEs) and serious adverse events (SAEs) was monitored, along with laboratory assessments (biochemistry, haematology, urinalysis), weight and body mass index (BMI). Levels of colistin in serum, urine and sputum were measured in patients receiving CDPI at visits 1, 2 and 6 and the time after inhalation was noted.
Other assessments included the following:
-
Patient's view of trial treatment (five-point scale assessing time to administer, ease of use, convenience).
-
Patient's preference (five-point scale, only in patients randomised to CDPI who therefore had experience of both study medications).
-
Quality of life (QoL) evaluated with the Cystic Fibrosis Questionnaire – Revised (CFQ-R, validated in European Union languages and for relevant age ranges).21
Before 24 weeks, intermittent assessments were made of safety, microbiology, use of other medications and other secondary endpoints. Pulmonary function was assessed by staff experienced in pulmonary function testing, blinded to the treatment being given.
Statistical analysis
At least 162 evaluable patients were required in each group to demonstrate non-inferiority of CDPI to TIS regarding relative change from baseline in FEV1% predicted after three cycles (24 weeks, including the 4-week periods off therapy). This was based on a two-group t test with a 0.05 two-sided significance level, and 16% SD. It was assumed that CDPI would be effective if the lower bound of the non-inferiority 95% CI was not more than −3%, based on the assumption that TIS was 6% better than placebo.22 Pre-planned subgroup analyses by age were also carried out. Secondary endpoints were of an exploratory nature.
The data did not follow a normal distribution and results were therefore evaluated using log-transformation analysis.
Secondary objectives were to compare changes in the microbiological sensitivity of P aeruginosa to study medication and to compare QoL measures.
The safety population consisted of patients who received at least one dose of medication. The intention-to-treat last observation carried forward (ITT LOCF) group included patients who had proven infection and at least one in-trial assessment with the last value evaluated. The ITT completers group included patients who, in addition, had a week 24 endpoint, having completed the study. The per protocol (PP) group comprised all randomised patients who had been exposed at least once to study medication, who were available for efficacy evaluation and who did not meet any violation criteria.
Results
A total of 380 patients were randomised: 187 CDPI and 193 TIS (figure 2). All except one patient received at least one dose of study medication and were included in the safety populations. The ITT group initially contained 374 patients; one patient dropped out immediately after baseline, therefore 373 were available for evaluation. Baseline demographic and clinical characteristics were similar between the two groups (table 1). There were 81 protocol violators; therefore the PP group contained 298 patients (figure 1).
Baseline patient demographic and clinical characteristics (intention-to-treat group)

Patient disposition. *One patient dropped out immediately following randomisation and did not receive treatment. CDPI, Colobreathe dry powder for inhalation; ITT, intention-to treat; PP, per protocol; TIS, tobramycin inhaler solution.
There was no significant difference in discontinuation rates between the groups: 32 and 21 patients respectively. AEs were the main reason for discontinuation in the CDPI group (18 patients).
Efficacy
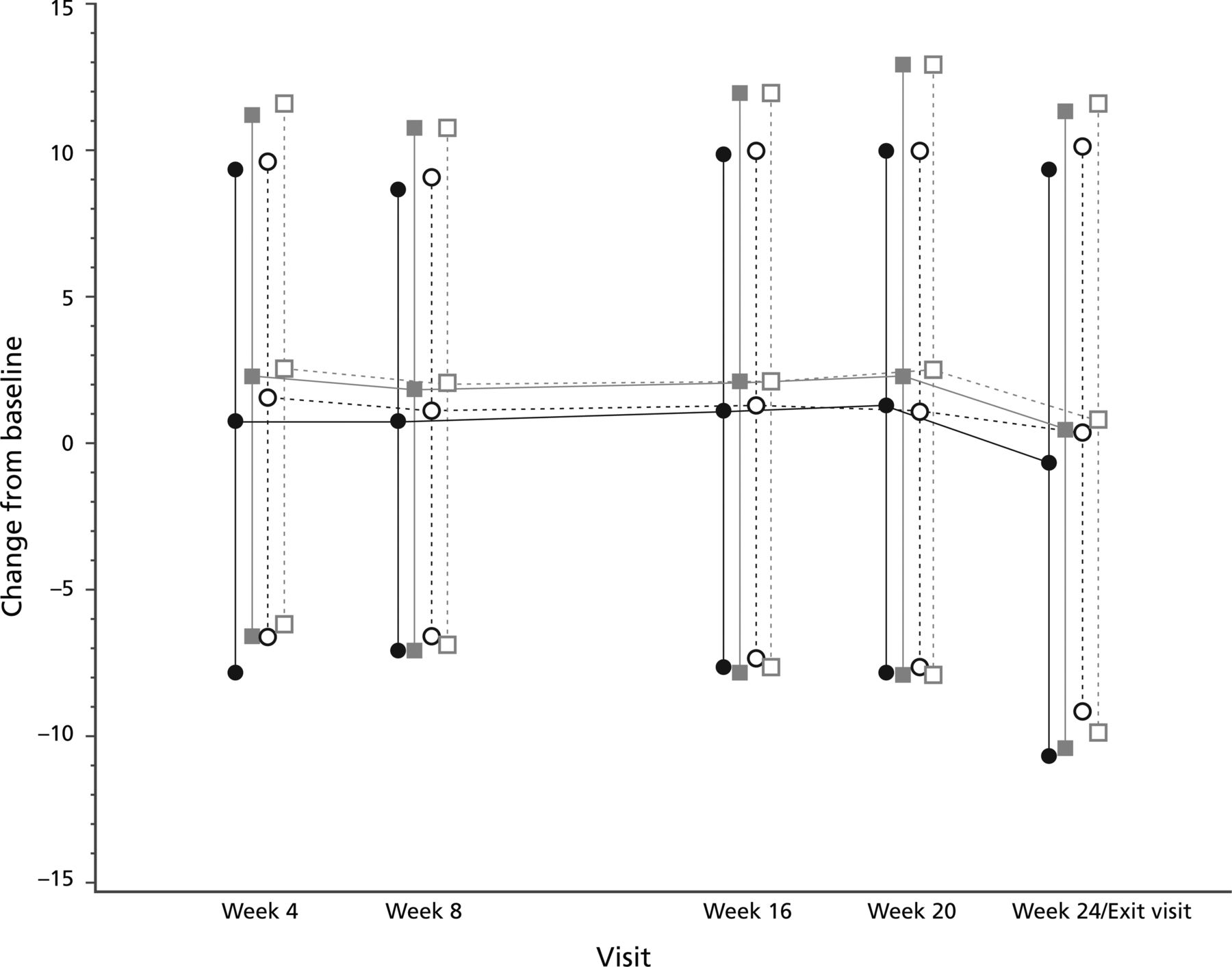
Mean changes in FEV1% predicted from baseline to week 24 were 0.964 for CDPI and 0.986 for TSI (table 2, figure 3). The adjusted mean difference between the groups in the change in FEV1% predicted at week 24 was therefore −0.98% (95% CI −2.74% to 0.86% (ITT LOCF)). The lower limit of the 95% CI (−2.71%, for patients who completed the study) was within the predefined 3% margin for treatment difference.

Forced expiratory volume in 1 s % predicted change from baseline (intention-to-treat population).  = Colobreathe dry powder for inhalation (CDPI) group;
= Colobreathe dry powder for inhalation (CDPI) group;  = tobramycin inhaler solution (TIS) group;
= tobramycin inhaler solution (TIS) group;  = CDPI completed patients;
= CDPI completed patients;  = TIS completed patients.
= TIS completed patients.
Changes in FEV1% predicted at week 24 (intention-to-treat population)
In the PP completers population, the adjusted mean difference between the groups in the change in FEV1% predicted at week 24 was −0.56% (95% CI −2.71% to 1.70%) (table 3).
Changes in FEV1% predicted at week 24 (per protocol population)
There was no statistically significant difference in the adjusted treatment difference for the change in FVC from baseline to week 24, for either the ITT (0.01 litre; 95% CI −0.09 to 0.10; p=0.886) or PP (−0.02 litre; 95% CI −0.12 to 0.08; p=0.697) populations. The adjusted treatment difference for the change in FEF25–75 was −0.12 litre/s (95% CI −0.23 to −0.01) in the ITT population (p=0.038) and −0.12 litre/s (95% CI −0.26 to 0.01) in the PP population (p=0.063) (see online supplementary table S1 for results of other pulmonary function tests).
In both treatment groups mean MIC50 values for colistin remained constant throughout the 24-week study period at 0.38 mg/litre, and ranged from 1.0 to 1.5 mg/litre for tobramycin (see table 2 for results of the sensitivity testing). Over the same period, in both groups the MIC90 values for colistin were 0.5–1.0 mg/litre, while those for tobramycin were 8.0–96.0 mg/litre, with the majority of tobramycin assessments being 12.00–16.00 mg/litre (see online supplementary table S2).
The proportion of colistin-resistant isolates in both groups was low (≤1.1%) (see online supplementary table S3). No increase was seen in the number or percentage of colistin-resistant isolates.
QoL measures
Although there were no statistically significant treatment differences in the change in overall QoL scores, the adjusted mean changes at week 24 favoured CDPI for treatment burden (p=0.091). This difference was significant at week 4 (p<0.001; figure 4). The study was not powered to detect differences in overall QoL. At that time no data existed that allowed the powering of a study for this outcome (see online supplementary tables S4 and S5 which provide QoL and patient ease of use data).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Change in Cystic Fibrosis Questionnaire Revised - treatment burden (intention-to-treat population: all ages).  = Colobreathe dry powder for inhalation (CDPI) group;
= Colobreathe dry powder for inhalation (CDPI) group;  = tobramycin inhaler solution (TIS) group.
= tobramycin inhaler solution (TIS) group.
Safety
The number of AEs was similar for both groups (table 4). There was a higher incidence of cough (75.4% vs 43.5%), throat irritation (45.5% vs 28.0%) and abnormal taste (62.6% vs 27.5%) in the CDPI group compared with the TIS group. Haemoptysis was below the 5% reporting level but was reported by more patients receiving CDPI (10.7%) than TIS (6.7%). Most AEs (2302/2426, 95%) were mild to moderate and the majority resolved without sequelae.
Most frequent adverse events by preferred term (≥5% of total number of events)
The incidence of treatment-related AEs was higher in the CDPI group (153/186 patients, 82.3% compared with 90/193, 46.6%), and discontinuations where the primary cause was an AE were also higher in this group (18/186 patients, 9.7%, compared with 3/193, 1.6%). A further six patients mentioned AEs as a reason for discontinuation, but not as the main reason. SAEs were higher in the TIS group (6.2% patients compared with 4.3%).
There were two deaths in the TIS group (lower respiratory tract infection related to underlying CF in both patients, additionally arterial thrombosis and multi-organ failure in one), both of which were thought to be unrelated to study medication.
There were no relevant changes in weight, BMI or growth in either group. There were no significant treatment-related changes in any haematological, biochemistry or urinalysis parameter.
In sputum, 171/451 (38%) samples had colistin levels ≥128 mg/litre, while the remainder had levels <128 mg/litre. Sputum levels were approximately 20 times the MIC of colistin. A total of 484 serum samples were available for testing. All but one were either just detectable or below the limit of detection (2 mg/litre). One sample had a level of >32 mg/litre. A total of 483 urine samples were available for assessment: the majority (377/483, 78%) of colistin concentrations were below 8 mg/litre, 76 samples were between 8 and 32 mg/litre, 18 between 32 and 64 mg/litre, 11 between 64 and 128 mg/litre and only one sample was above 128 mg/litre.
Other assessments
The Turbospin device used with CDPI was rated ‘very easy to use’ by 51.9% of the CDPI group, whereas only 9.9% of patients in the TIS group rated their device as ‘very easy to use’ (ITT). The proportion in each group who rated their device as ‘very easy or easy to use’ was 90.7% vs 53.9% respectively (p<0.001) (see online supplementary tables).
Patients assigned to CDPI compared CDPI (Colobreathe plus Turbospin device) to their pre-randomisation experience with TIS (TOBI with a PARI LC Plus nebuliser plus compressor). In the ITT population 65.6% of patients preferred the CDPI treatment. In the 6–12-year age group 80.6% of children preferred the CDPI treatment mode.
Discussion
Our study demonstrated that colistimethate sodium, delivered as dry powder for inhalation, is non-inferior to TIS in the treatment of chronic P aeruginosa infection in patients with CF as measured by change in FEV1% predicted at 24 weeks. TIS is effective relative to placebo in maintaining lung function,23 with FEV1% being a predictor for survival in CF,24 therefore it can be surmised that CDPI would have a similar outcome.
Regular monitoring of the variability of FEV1 was carried out to check whether the SD was within 16%, as had previously been documented in patients with CF, as this variability would impact the CI.23 In fact, in this study, the SD of FEV1 lies within 13%, reflecting rigorous adherence to inclusion criteria.
There was no change in the susceptibility of P aeruginosa isolates to colistin in either group. Since the first publication in 198525 suggesting inhaled nebulised colistimethate sodium in patients with CF colonised with P aeruginosa, there has been no significant increase in resistance of P aeruginosa isolates, despite widespread use of colistimethate sodium for over 20 years.26
This study confirmed the good safety profile of CDPI with a similar range of AEs in both groups, except for cough and abnormal taste which were more frequent in patients receiving CDPI. This is typical of inhaled dry powder therapies, as inhalation of these products causes mild cough, throat irritation and unpleasant taste, due to deposition in the oropharynx.27 The inspired payload of colistimethate sodium, 125 mg of powder, is unusually large and is likely to contribute to the irritant effect. Indeed we observed more withdrawals due to AEs in the CDPI group. However, this is not surprising as the TIS group were by definition TIS tolerant, having received at least two cycles in the pre-randomisation phase. Detailed run-in data were not collected but the majority of patients had received TOBI for 1–2 years (median 2 years). Most events diminished after 28 days when they were similar to the levels seen with TIS. A long-term study is necessary to monitor possible long-term effects.
One suggested advantage of inhaled therapies is that they deliver a higher fraction of drug to the bronchopulmonary system, although part is inevitably lost through impact with tissues in the mouth and pharynx. In this study the levels were measured using a single-reference microbiological assay, the only available validated assay at the time, which is now thought to lack sensitivity and precision (lower limit of quantification=0.5 µg/ml). However, the results do confirm previously reported findings. An ongoing study using improved assay techniques will be reporting soon.
Inhalation time with older nebulisers is up to 20 min twice a day, although newer models are much quicker. A clean environment is required to prepare the solution, and regular cleaning is required to avoid the apparatus becoming a source of infection.28 The compressor also requires regular maintenance.
Users of inhaled dry powder devices should be able to take an effective dose of one capsule in around 1 min. Administration of a dose from the TOBI Podhaler, the dry powder formulation of tobramycin, requires 6–7 min as four capsules are inhaled for each dose;27 Colobreathe only requires one capsule per dose. No special storage conditions are required for the Colobreathe capsules. The Turbospin device is cleaned with a dry wipe and does not require washing. It is discarded after 28 days’ use, but remains effective during this period without any clogging of the delivery mechanism.
Patients in this study rated CDPI more highly for ease of use. Although adherence and the possible impact on long-term efficacy were not measured in this study, other studies have shown that improved convenience and reduced complexity of treatment can have a positive impact on adherence.14 ,29 ,30
Conclusion
CDPI has been shown to be effective and is non-inferior to TIS over 24 weeks of treatment in patients with CF chronically infected with P aeruginosa. Emergence of resistance of P aeruginosa to colistin was not seen. The QoL domain, burden of treatment, favoured CDPI over TIS at week 4. Overall, CDPI was well tolerated.
Acknowledgments
We are grateful to Jude Douglass, Healthcom Partners, Oxford for linguistic and editorial assistance in the preparation of the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online tables
Footnotes
-
Correction notice This article has been corrected since it was published Online First. The Results section of the abstract have been updated to read: After logarithmic transformation of data due to a non-normal distribution, adjusted mean difference between treatment groups (CDPI vs TIS) in change in forced expiratory volume in 1 s (FEV1% predicted) at week 24 was -0.98% (95% CI -2.74% to 0.86%) in the intention-totreat population (n=373). The Efficacy section has been updated to read: The adjusted mean difference between the groups in the change in FEV1% predicted at week 24 was therefore -0.98% (95% CI -2.74% to 0.86% (ITT LOCF)). The lower limit of the 95% CI (-2.71%, for patients who completed the study). The legend for Figure 4 has been amended.
-
Contributors All authors contributed to the interpretation of data and to the development and editing of the manuscript.
-
Funding Funding for the study was provided by Forest Laboratories UK, Dartford. The full trial protocol can be inspected on application to the sponsor.
-
Competing interests AS received investigator fees and honoraria for presentations related to the study. CH is a consultant to Forest Laboratories UK Ltd. GD is a consultant to Forest Laboratories UK Ltd. MHG is an employee of Forest Laboratories UK Ltd., owns shares in Forest Laboratories Inc, and is named on the EU patent for Colobreathe as one of the inventors.
-
Ethics approval Independent Ethics Committees at each institution.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement The original clinical study report for this study, along with laboratory reports, are available for inspection on request. Please contact the study sponsor.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/