Article Text

Abstract
Matrix metalloproteinase-9 (MMP-9) is increased in a number of pathological lung conditions, where the proteinase contributes to deleterious remodelling of the airways. While both lung cancer and COPD are associated with increased MMP-9 expression, the cellular and molecular drivers of MMP-9 remain unresolved. In this study, MMP-9 transcript measured within the tumour region from patients with non-small-cell lung cancer (NSCLC) and coexisting COPD was found to be uniformly increased relative to adjacent tumour-free tissue. MMP-9 gene expression and immunohistochemistry identified tumour-associated neutrophils, but not macrophages, as a predominant source of this proteinase. In addition, PTEN gene expression was significantly reduced in tumour and there was evidence of epithelial MMP-9 expression. To explore whether PTEN can regulate epithelial MMP-9 expression, a small interfering (si)RNA knockdown strategy was used in Beas-2B bronchial epithelial cells. PTEN knockdown by siRNA selectively increased MMP-9 expression in response to lipopolysaccharide in a corticosteroid-insensitive manner. In summary, tumour-associated neutrophils represent an important source of MMP-9 in NSCLC, and loss of epithelial PTEN may further augment steroid-insensitive expression.
- COPD ÀÜ Mechanisms
- Lung Cancer
- Airway Epithelium
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
The global prevalence of lung cancer and COPD will continue to rise, as smoking consumption is yet to peak in developing regions. Mutagens and free radicals in cigarette smoke damage and transform the lung epithelium, leading to emergence of a squamous metaplasia phenotype that correlates with the severity of COPD.1 In addition to mutagens in tobacco, shared molecular drivers will contribute to the emergence of lung cancer in people with COPD.2 Chronic inflammation is a recognised enabling characteristic in cancer, where inflammatory and metaplastic cells secrete a milieu of growth factors and cytokines to drive remodelling processes. This inflammatory environment can also promote increased expression of proteases that degrade extracellular matrix and basement membrane, which is required for migration of leucocytes and cancer cells. One such proteinase known to be elevated in COPD is matrix metalloproteinase-9 (MMP-9), where neutrophils are an important source of corticosteroid-resistant MMP-9 activity.3 In addition to leucocytes, the number of basal epithelial cells positive for MMP-9 has been found to be elevated in COPD.4
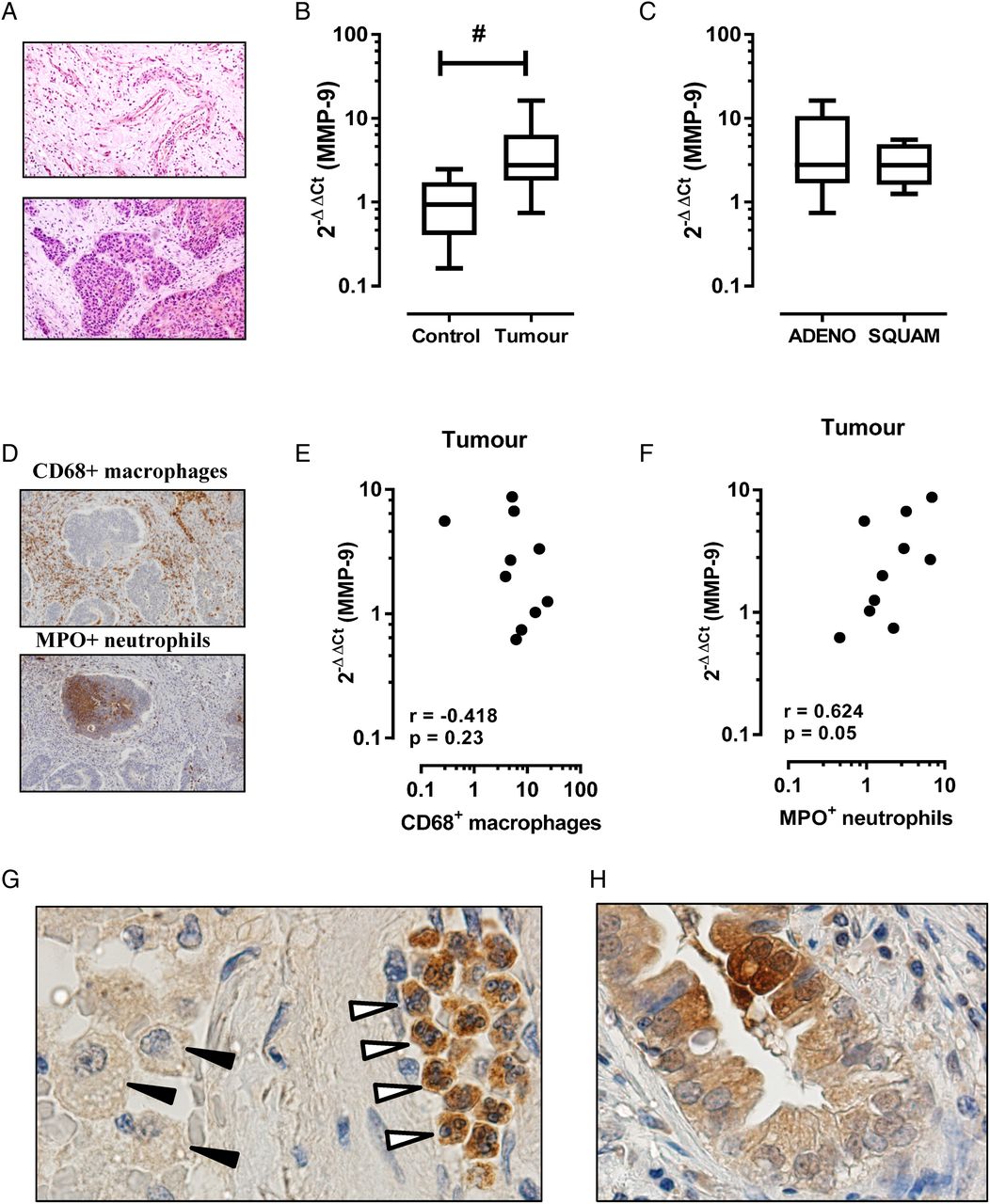
MMP-9 is also known to be increased in lung cancer, where it can play multiple roles in tumour progression including angiogenesis and establishment of a metastatic niche. As in COPD, the cellular source of MMP-9 is likely to be multifactorial in lung cancer including inflammatory leucocytes and tumour cells of epithelial origin. In this study, we show that MMP-9 transcript expression is increased in the tumour region relative to the adjacent tumour-free region. Figure 1A is a representative tissue section of a tumour-free (figure 1A, top panel) and intratumour (figure 1A, bottom panel) region from a patient with COPD and histologically confirmed squamous cell carcinoma. MMP-9 transcript expression was significantly higher in the tumour sample (figure 1B; mean±SEM 4.6±1.5-fold increase, p<0.05) relative to the adjacent control tissue (1.1±0.25-fold increase). Stratification by tumour histology revealed no significant difference between tumour subtypes (figure 1C, adenocarcinoma 5.5±2.4-fold increase vs squamous cell carcinoma 3.1±0.9-fold increase). The percentage area of positive staining for CD68+ macrophages and MPO+ neutrophils revealed no significant difference in tissue accumulation of either leucocyte, although there was a trend towards increased numbers of tumour-associated neutrophils (2.7±0.7% positive area) relative to adjacent control tissue (1.4±0.2% positive area). Spearman correlation revealed no association between MMP-9 expression and macrophage staining (figure 1E, r=−0.42, p=0.23), whereas a closer association between MMP-9 expression and tumour-associated neutrophils was observed (figure 1F, r=0.63, p=0.05). Consistent with this finding, immunohistochemistry for MMP-9-positive cells using serial tumour sections identified intensely staining neutrophils in the tumour tissue relative to weakly staining macrophages (figure 1G). MMP-9 immunoreactivity in cells of epithelial origin was also detected in tumour sections (figure 1H).
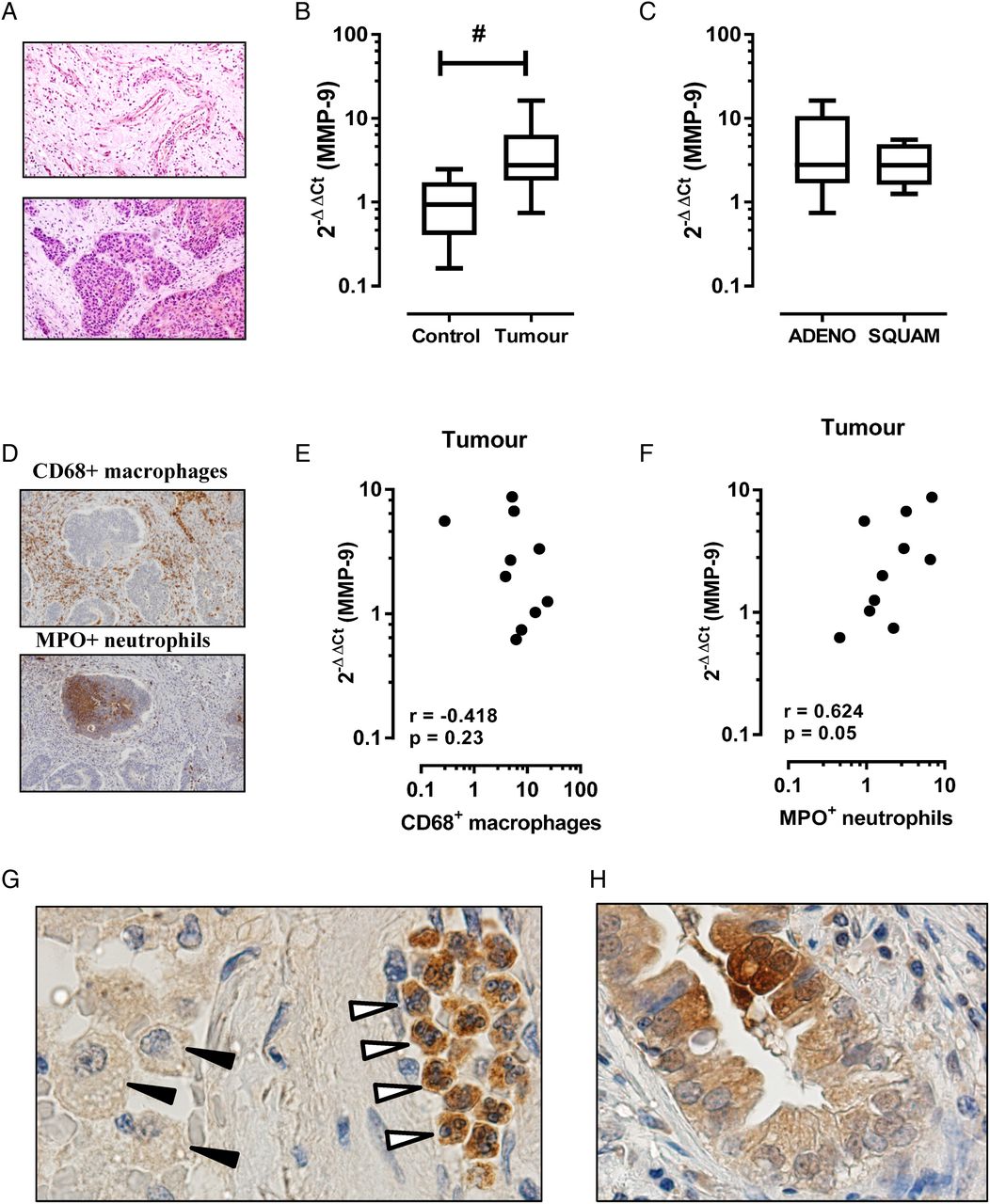
Matrix metalloproteinase-9 (MMP-9) transcript is increased in the tumour region in patients with COPD. (A) Representative H&E-stained tissue section from formaldehyde-fixed paraffin-embedded blocks of tissue from the tumour site (bottom panel) and the adjacent parenchyma that was histologically free of tumour (top panel). (B) MMP-9 levels were expressed as fold change relative to individual adjacent control sample (n=10; #Wilcoxon signed rank test, p<0.05). (C) Levels of MMP-9 grouped on the basis of non-small-cell lung cancer tumour subtype showed no difference in expression. ADENO, adenocarcinoma; SQUAM, squamous cell carcinoma. (D) Representative immunostain of tumour sections for CD68 (top panel) and MPO (bottom panel). (E) Spearman correlation demonstrates no association between CD68+ macrophages and MMP-9 expression, whereas (F) MPO+ neutrophils demonstrated a closer relationship with MMP-9 in tumour sections. (G) Representative tumour section stained for MMP-9 identified intense immunoreactivity in tumour-associated neutrophils (open arrows), but not macrophages (black arrows). (H) There was also evidence of MMP-9 immunoreactivity in cells of epithelial origin.
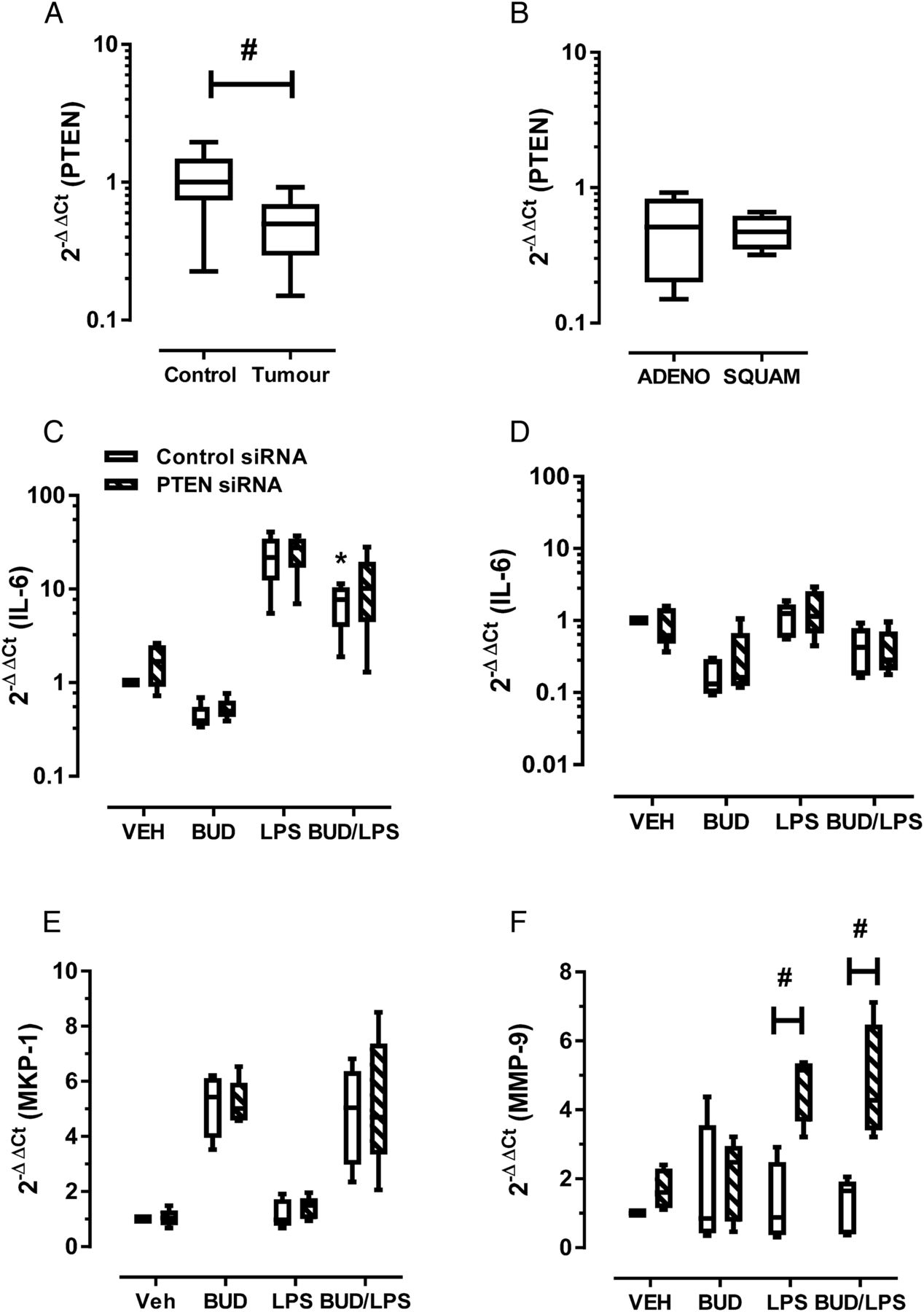
Since tumour cells can also express MMP-9 in NSCLC,5 we investigated mechanisms by which cells of epithelial origin could contribute to increased MMP-9. In our analysed samples, PTEN gene expression was 0.5±0.08-fold lower in the tumour region (figure 2A, #p<0.05). Consistent with MMP-9 expression, no significant difference in PTEN levels was observed between NSCLC subtypes (figure 2B). Loss of PTEN increases phosphoinositide-3-kinase (PI3K)/Akt pathway activation in smokers with dysplastic regions, suggesting that PI3K activation is an early event in tumorigenesis6 and PTEN was progressively downregulated in COPD epithelium.7 To investigate whether PTEN can contribute to regulation of MMP-9 expression, siRNA was used in bronchial epithelial Beas-2B cells to reduce PTEN expression by 50–70% (see online repository for methods and additional data).
{kind=link}
{kind=link}
Effect of epithelial PTEN knockdown (KD) on inflammation and matrix metalloproteinase-9 (MMP-9) expression. (A) PTEN levels were significantly reduced at the tumour site relative to tumour-free control section (#Wilcoxon signed rank test, p<0.05). (B) Levels of PTEN grouped on the basis of non-small-cell lung cancer tumour subtype demonstrated no difference in expression. ADENO, adenocarcinoma; SQUAM, squamous cell carcinoma. (C) Beas-2B cells transfected with control or PTEN siRNA were stimulated with 100 ng/mL lipopolysaccharide (LPS) and 10−7 M budesonide (BUD). Interleukin-6 (IL-6) levels were measured at (C) 3 hours and (D) 48 hours. LPS-induced IL-6 expression at 3 hours was significantly reduced in control but not PTEN KD cells (*p<0.05, Friedman test with Dunn’s correction of LPS vs BUD-LPS). (E) As a control for steroid exposure, mitogen-activated protein kinase (MAPK) phosphatase-1 (MKP-1) levels were determined 3 hours after treatment; no difference between control and PTEN KD cells was seen. (F) At the 48 hour time point, MMP-9 expression was determined, demonstrating a significant increase in LPS-stimulated PTEN KD cells but not control cells, and this response was not inhibited by budesonide (#p<0.05, two-way analysis of variance and Bonferroni multiple comparison post hoc test).
supplementary data
Control and PTEN knockdown (PTEN KD) cells were stimulated with the bacterial agonist lipopolysaccharide (LPS), which increased interleukin-6 (IL-6) levels approximately 20-fold (figure 2C) at 3 hours, with expression returning to baseline by 48 hours (figure 2D). These time points were chosen to represent the peak (3 hours) and resolution (48 hours) of inflammatory responses. No significant difference was observed between control and PTEN KD cells. Budesonide significantly reduced peak LPS-induced IL-6 levels in control cells (figure 2C; 70% reduction; analysis of variance (ANOVA), *p<0.05 control cells LPS vs BUD-LPS). Budesonide proved to be less effective in reducing peak IL-6 levels in PTEN KD cells (figure 2C; 54% reduction; p>0.05 PTEN KD cells LPS vs BUD-LPS). Since mitogen-activated protein kinase (MAPK) phosphatase-1 (MKP-1) contains consensus glucocorticoid response elements in its promoter, this gene was measured as a control for steroid exposure. Budesonide-induced levels of MKP-1 were not altered in PTEN KD cells at 3 hours, hence steroid receptor engagement and nuclear translocation is maintained (figure 2E).
Preliminary assessment of MMP-9 expression at 3 hours demonstrated no increase in MMP-9 levels in either control or PTEN KD cells (data not shown). At the later 48 hour time point, LPS stimulation of control cells did not significantly alter baseline MMP-9 expression. In contrast, PTEN KD cells responded to LPS by increasing MMP-9 expression (figure 2F; 4.7±0.5-fold increase; two-way ANOVA #p<0.05, control vs PTEN KD cells). LPS-induced expression of MMP-9 was not inhibited by budesonide in PTEN KD cells (figure 2F; 4.7±0.8-fold increase; BUD-LPS treated control vs PTEN KD cells, two-way ANOVA #p<0.05). The promoter region of human MMP-9 contains binding sites for multiple transcription factors. We have previously shown that the activator protein-1 (AP-1) was markedly increased in LPS-stimulated Beas-2B cells;8 hence AP-1 requires synergistic cooperation with multiple transcription factors to stimulate MMP-9 expression. IL-6 interacts with glycoprotein 130 and activates the JAK/Stat pathway. PTEN is an important negative regulator of Stat3, and functional cooperation of the Stat3 and AP-1 transcription factors plays a crucial role in transcription of the MMP-9 gene through engagement of juxtaposed promoter elements.9 Hence, loss of PTEN may result in enhanced Stat3–AP-1 interactions to sustain MMP-9 expression.
In summary, tumour-associated neutrophils in NSCLC were most strongly associated with MMP-9 expression in our study. Neutrophils can be transformed by the malignant microenvironment, leading to divergent phenotypes that release proangiogenic factors such as MMP-9.10 We also propose that reduced expression of epithelial PTEN may activate an alternative transcriptional network to stimulate steroid-insensitive epithelial expression of MMP-9. Hence, novel strategies aimed at reducing tumour-associated neutrophils and restoring epithelial PTEN expression should be considered.
Footnotes
Contributors AV and SB: responsible for generation of hypothesis and experimental design of all experiments; they conducted most of the experiments and wrote and revised the manuscript. HJS, GA, MT, LBI, RV and DS: contributed to the experimental design and data analysis and contributed to manuscript preparation.
Funding National Health and Medical Research Council; Australian Research Council.
Competing interests None declared.
Ethics approval Royal Melbourne Hospital Human Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.