Article Text

Statistics from Altmetric.com
Alpha-1-antitrypsin deficiency was first identified in 1963, together with its association with the early onset of severe lower zone emphysema.1 Although the mechanisms for the development of emphysema were not clearly understood, it was reasonably assumed that the alveolar destruction was a direct consequence of the release of neutrophil elastase which was then able to digest lung connective tissue because of the low concentrations of α1-antitrypsin. This led to the concept and development of augmentation therapy2 which was shown to restore both the serum and alveolar concentration2 ,3 of α1-antitrypsin to a level thought to be adequately protective. This represented a relatively simplistic approach and augmentation therapy became an accepted form of management, initially in the USA, but subsequently in Germany and, more recently, for some patients in several other European countries. The efficacy of such an approach remains contentious. For many years it has been thought that an appropriate clinical trial would not be feasible in view of patient numbers required and the expense of designing and monitoring such a trial. This has led to recent editorials raising the issue of whether an appropriate clinical trial will ever be conducted,4 ,5although most clinicians and research workers remain firmly supportive of such a trial.
In 1996 the WHO held a meeting of world experts and concluded that many questions still needed answering, including the nature of the lung pathology of α1-antitrypsin deficiency, the relative impact of deficiency on disease development, and rationalisation of the standards of care, and concluded that further research was necessary into the pathological processes in order to facilitate the design of adequately powered controlled clinical trials.6 This review addresses the more recent developments in α1-antitrypsin deficiency and outlines the potential methodologies that may enable the implementation of such a trial to clarify the efficacy of augmentation therapy.
Epidemiology
Alpha-1-antitrypsin deficiency is classically associated with the early onset of severe basal emphysema, but is also associated with the development of cirrhosis, primary carcinoma of the liver and the vasculitides. All these decrease life expectancy although the pulmonary condition dominates, as emphasised by the additive effect of cigarette smoking on mortality in patients with deficiency.7However, more recent epidemiological data from Sweden have indicated that airflow obstruction can develop even in non-smokers. In longitudinal studies based on their extensive national data base, Pittulainen and colleagues8 confirmed that lung function in non-smokers was well maintained until middle age. However, over the age of 50 a proportion of the deficient subjects developed increasing airflow obstruction which was shown to be related to a history of wheezing and occupational exposure to irritants. This raises the possibility that asthma may be a predisposing factor to the development of permanent airflow obstruction in non-smokers with α1-antitrypsin deficiency and supports one of the WHO recommendations that all adults with asthma should be tested for deficiency.6 Whether this, other volatile chemicals, and pollution within the environment play a role in the development of airflow obstruction in α1-antitrypsin deficiency remains to be determined.
Pathogenesis
The early concept of the development of emphysema in deficient subjects was based on a simple “balance” theory where the amount of elastase released in the lung exceeded the amount of α1-antitrypsin present in deficient subjects. The net result was persistence of elastase activity leading to lung destruction and the pathological changes of emphysema. However, recent elegant experiments involving neutrophil cell biology have clarified the mechanism involved. The studies also provide data to explain why subjects with partial deficiency (such as MZ heterozygotes) or moderate deficiency (such as SZ heterozygotes) do not appear at increased risk for the development of emphysema9 ,10 despite significantly reduced concentrations of α1-antitrypsin in the plasma (and, by implication, in the lung).
Alpha-1-antitrypsin is predominantly a serum protein that enters the lung by passive diffusion. Early physiological studies showed that the endothelium is relatively permeable with interstitial protein concentrations approximately 80% of that present in plasma.11 On the other hand, the epithelium presents a relatively impermeable barrier to protein movements such that concentrations of proteins within the epithelial lining fluid are approximately one tenth of that in plasma.11 It is generally believed that destruction of lung elastin in the interstitial space is the key to the development of emphysema.12 Thus, the concentration of proteinase inhibitors in this compartment is critical in the protection of elastin and this will be broadly similar to the plasma concentration.
Neutrophil elastase is stored within the azurophil granule of the mature polymorphonuclear leucocyte. These granules are exocytosed during activation of the cell and their protease content is released in active form and diffuses gradually away from the granule. The concentration of elastase within these granules is approximately 5 mM and thus over two orders of magnitude higher than the normal concentration of α1-antitrypsin within the plasma (30 μM) and hence within the interstitium (24 μM). This major disparity in concentration explains why neutrophils are able to digest connective tissue even when bathed in supernormal concentrations of α1-antitrypsin. Liou and Campbell13described this process as quantum proteolysis and developed a mathematical model to determine the concentration of elastase as it diffuses away from the azurophil granule. They argued that the enzyme would retain its ability to digest connective tissue until its concentration had dropped sufficiently to equal that of the surrounding inhibitors. Subsequent experiments with normal neutrophils produced experimental data to support this theoretical concept.14The authors were able to demonstrate that the concentration of elastase, as it diffuses away from the neutrophil, follows an exponential curve. Initially, the concentration falls rapidly in close proximity to the granule. However, concentrations of elastase below 10 μmol are retained over a wide area. The implication of this observation is that the elastase concentration drops rapidly to equal the likely concentration of α1-antitrypsin for most patients with normal or heterozygote deficiency. The proteolytic damage would therefore be limited to an area close to the granule even for subjects with partial deficiency. However, the only common phenotype with a serum α1-antitrypsin concentration below 10 μmol is the Pi Z phenotype which has an increased risk for development of rapidly progressive emphysema. The mean concentration of α1-antitrypsin in the plasma of these subjects is 5 μM, which theoretically results in an interstitial concentration of 4 μM (see above) and is thus well below the threshold of the exponential part of the elastase dilution curve. These theories and observations, together with the increased neutrophil recruitment to the lung of α1-antitrypsin deficient patients (thought to be related to excess leukotriene B4 generation by macrophages), explains the particular susceptibility of deficient subjects to develop extensive and rapidly progressive lung destruction. The mechanisms involved in this process are summarised in fig 1.
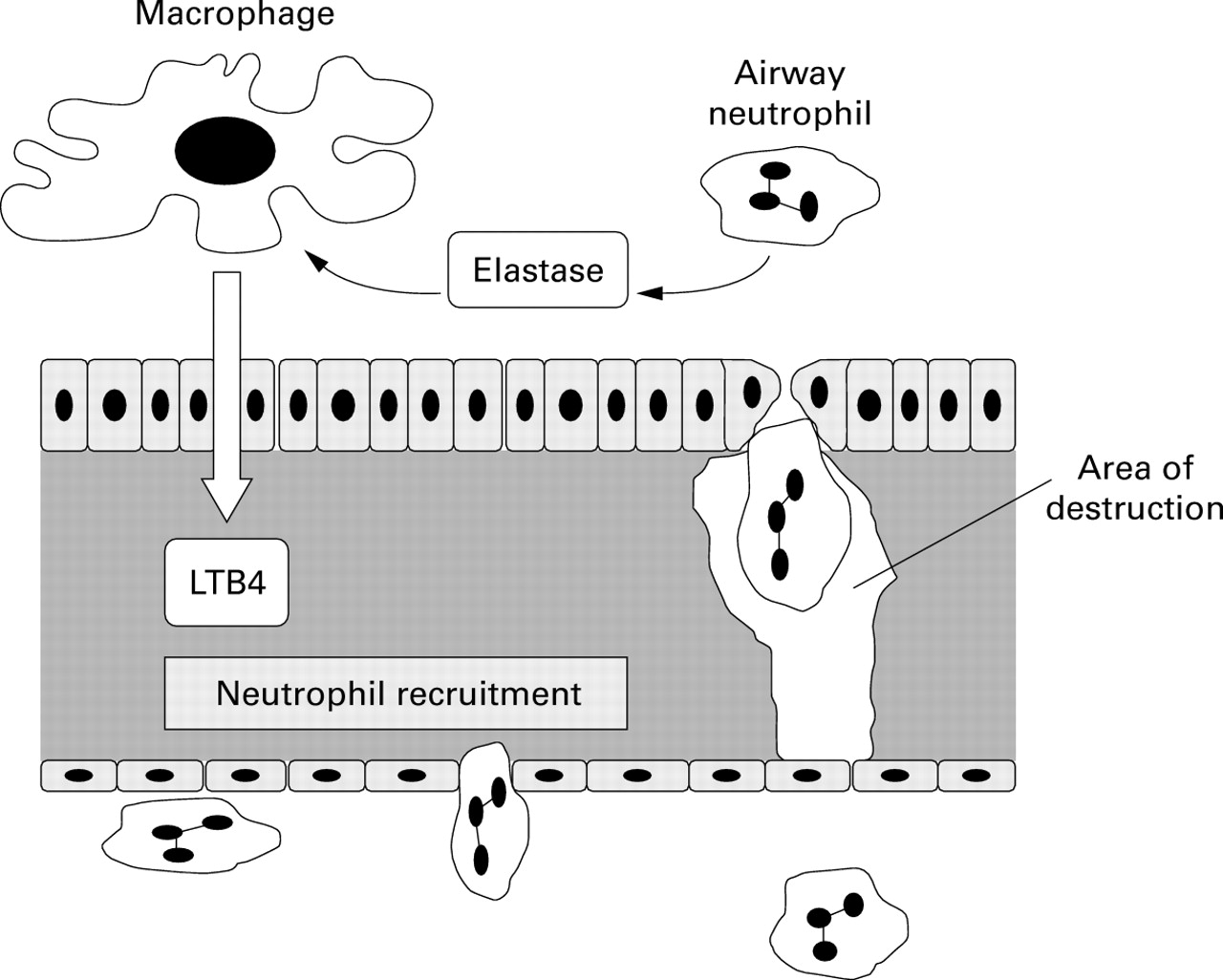
Suggested pathogenic mechanism for lung destruction in α1-antitrypsin deficiency. Activation of airway neutrophils leads to the release of neutrophil elastase which remains active because of a reduced inhibitory capacity as a direct consequence of α1-antitrypsin deficiency. The elastase stimulates macrophages to release the chemoattractant leukotriene B4 (LTB4) which leads to further neutrophil recruitment. Recruitment of neutrophils through the interstitium causes connective tissue destruction (particularly elastin) and the area of destruction is enhanced again as a direct consequence of α1-antitrypsin deficiency. The net result is perpetuation and amplification of the cycle of events and tissue destruction.
Understanding this process has major implications for the design of clinical trials. Intravenous therapy restores and retains the circulating concentration above 11 μM. This will predictably lead to a basal interstitial α1-antitrypsin concentration of 8–9 μM which would be predominantly protective (as for the SZ phenotype). On the other hand, approaches to deliver α1-antitrypsin to the alveolar surface could prove less effective unless the concentration that can be achieved is sufficiently high to allow adequate quantities of α1-antitrypsin to pass through the tight epithelial layer into the interstitium and raise the concentration to a protective level.
However, it is possible that a strategy of delivery of α1-antitrypsin to the airway could also have a beneficial effect even if the protein does not reach the interstitium. This is based on the concept that, if neutrophil recruitment to the lung can be reduced, then protection of the interstitium becomes less critical. Hubbard and colleagues had provided evidence that excess neutrophil recruitment to the lung was based on the release of leukotriene B4 (LTB4) by alveolar macrophages due to the presence of uninhibited elastase activity in the alveolar space.15 Direct augmentation of α1-antitrypsin in the alveolar region would be predicted to inhibit any elastase released, thus switching off LTB4 production by the macrophage and hence subsequent neutrophil recruitment. The reduced neutrophil traffic would lead to less elastin degradation and hence cessation or modulation of the progression of emphysema (fig 1). Clearly, this concept is testable by appropriately designed research studies and should be included in clinical trial design.
Role of augmentation therapy
As indicated above, augmentation therapy seems a logical step to take in the management of patients with α1-antitrypsin deficiency. However, cessation of smoking clearly remains critical and identification and hence avoidance of other pollutants or risk factors becomes equally important. Indeed, there is currently a combined ERS/ATS task force addressing the current standards of care for α1-antitrypsin deficiency and the guidelines should be published within the next year.
In a previous editorial Stoller5 reviewed historical data from two European countries, one of which had augmentation therapy available,16 and the final data from the NHLBI registry in the USA17 comparing patients who had or had not received augmentation therapy. Both studies provided evidence that subjects who received augmentation therapy retained their lung function better, although in the latter study this was confined to subjects with moderate impairment. More interestingly, the mortality rate appeared to be reduced in American subjects who were able to receive augmentation therapy.17 Clearly, neither study was a controlled trial and the fact that the groups were not matched for either country, health care insurance, or exposure to health care workers still leaves doubts concerning efficacy. More recently a combined study between Holland and Denmark conducted over three years showed no effect of augmentation on decline in lung function.18 This was a double blind controlled study although unfortunately the study was insufficiently powered to demonstrate an effect on lung function (28 versus 28 patients). Of interest, however, this study suggested that augmentation prevented deterioration in the degree of emphysema as assessed by CT scanning, although this just failed to achieve conventional levels of statistical significance (p<0.07). Nevertheless, since α1-antitrypsin deficiency is specifically thought to be associated with the development and progression of emphysema, this observation is likely to be of major importance. It raises the critical issue of whether progression of the CT scan should be a primary outcome measure and, if so, this would reduce the number of patients to 130 and the length of study required to three years to demonstrate efficacy.18
Future clinical trials
Demonstrating the efficacy of augmentation therapy requires appropriate phase II (proof of principle) studies and adequately powered controlled clinical trials with accepted primary and secondary outcome measures.
PHASE II STUDIES
In the past, phase II studies have been confined to demonstrating the restoration of α1-antitrypsin concentration and function. Whereas this is clearly important, it does not demonstrate an effect on the central pathogenic progresses involved in disease development, progression, and health status.
Inflammatory markers
Studies of bronchoalveolar lavage fluid19 and sputum20 from the large airways have shown that inflammation is increased in α1-antitrypsin deficiency. This provides a surrogate of the processes thought to be central to the development of disease in α1-antitrypsin deficiency. It should be possible to determine whether α1-antitrypsin augmentation influences both the biochemical and cellular processes in the lung. Indeed, if the theory proposed by Hubbardet al 19 concerning the perpetuation of neutrophil recruitment in α1-antitrypsin deficiency is correct, augmentation therapy should lead to a reduction in LTB4 release within the lung and subsequently a reduction in neutrophil numbers as recruitment decreases. Preliminary studies are underway to determine whether these effects are of importance.
Bacterial colonisation
Many patients with α1-antitrypsin deficiency have evidence of chronic bronchial disease with bacterial colonisation.20 Recent data have suggested that, although this is considered to be a benign process in the stable clinical state, it may be a major contributing factor to inflammation in the airway in some patients.21 Indeed, studies have shown that patients with α1-antitrypsin deficiency have a greater degree of inflammation in the presence of bacterial colonisation than in non-deficient subjects.20 Bacterial colonisation may be facilitated by epithelial damage, reduction in ciliary beating, and mucus hypersecretion. All these are features that can be induced by inflammation with uncontrolled neutrophil elastase activity.22 Alpha-1-antitrypsin augmentation in deficient subjects would increase the inhibitory capacity of the lung secretions and may protect these important defence mechanisms. This could reduce colonisation and airway inflammation. Whether this bacterial colonisation is responsible for disease progression, morbidity, or recurrent exacerbations remains uncertain. Nevertheless, short term studies of the effect of augmentation on airway colonisation, mucociliary clearance, or inflammation may also form the basis of sound phase II studies.
Biochemical markers
Biochemical markers of progression of lung disease have been extensively investigated as tools to determine efficacy for phase II studies but have so far proved too insensitive. Destruction of lung elastin is thought to be central to the development of emphysema but monitoring elastin breakdown products such as desmosine and iso-desmosine has not proved helpful. Levels are raised in chronic obstructive pulmonary disease (COPD)23 but not in α1-antitrypsin deficiency,24 although this may reflect methodological differences. Snider and colleagues, however, have indicated that augmentation therapy reduced the quantity of these breakdown products excreted in the urine in a limited study involving only two patients.25 More recently smoking has also been shown to lead to the release of breakdown products of lung collagen in experimental animals.26 Augmentation with α1-antitrypsin reduced this and it may be that further studies of other connective tissue breakdown products could prove useful in determining the influence of augmentation therapy. Clearly, further studies are indicated but it is unlikely that such a marker would be accepted as indicative of clinical efficacy in the absence of other pathological or physiological support.
CONTROLLED TRIALS
The efficacy of treatments for patients with chronic lung disease has focused on forced expiratory volume in one second (FEV1) as the primary outcome measure. Indeed, although a reduction in FEV1 in emphysema is largely dependent on dynamic airways collapse, treatments that can acutely increase the FEV1 are often prescribed even in the absence of clear physiological improvement. However, the FEV1 is also considered the outcome measure of choice for treatments designed to prevent progression of chronic lung disease. Whereas this may be reasonable, measurement of FEV1 is effort dependent and can vary from day to day even in a stable patient.
Thus, clinical trials designed to assess modification of FEV1 require large numbers of patients followed for several years.
In the case of chronic lung disease in general, and the emphysema of α1-antitrypsin deficiency in particular, alternative outcome measures for long term disease modification should be considered. These might include tests of dynamic airways collapse such as the maximum expiratory flow (MEF25–75), air trapping, flow limitation, exercise capacity, or more specific tests of alveolar function such as the carbon monoxide transfer coefficient (Kco). However, the most specific way of assessing the extent of emphysema is with computed tomographic (CT) scanning as studies have shown that analysis of the scans correlates well with the degree of emphysema in pathological specimens.27 CT scanning relates well to tests of airflow obstruction and gas transfer in patients with emphysema28 and preliminary studies have shown that, in patients with α1-antitrypsin deficiency, progression of emphysema assessed by CT scanning is detectable in a small number of patients over as little as 12 months.29Indeed, the recent Dutch/Danish controlled trial would suggest that CT scanning is a sensitive way of determining the efficacy of replacement therapy18 and should clearly be included as a secondary, if not primary, outcome measure in controlled clinical trials.
Exacerbations
Exacerbations are a common occurrence in patients with chronic lung disease and such episodes are associated with a worsening of quality of life which may take some months to recover.30Patients with α1-antitrypsin deficiency often experience regular exacerbations of their lung disease and, indeed, the inflammation associated with these episodes is much more severe than that seen in corresponding patients without deficiency.31This might be expected since α1-antitrypsin is an acute phase protein and in normal subjects the serum concentration rises during exacerbations and, together with increased protein leakage, leads to a corresponding increase in lung α1-antitrypsin levels. In subjects with severe deficiency there is virtually no acute phase rise in serum concentration and, although the concentration rises in the lung as a result of increased inflammation, it remains well below that seen for non-deficient subjects.31 The normal physiological response of α1-antitrypsin during such episodes may help to control the increased elastase released by the greater neutrophil recruitment during the exacerbation, thereby containing and curtailing the degree of inflammation. This raises the possibility that intravenous boluses of α1-antitrypsin may play a key role in the modulation of exacerbations, although whether this will influence long term progression remains currently uncertain. Nevertheless, the number of exacerbations and exacerbation days have been used in other studies to demonstrate clinical efficacy of therapeutic intervention32 and such a clinical design is feasible in α1-antitrypsin deficiency. In other studies as few as 30 patients have been necessary to prove efficacy and this would clearly be possible in α1-antitrypsin deficiency.
Health status
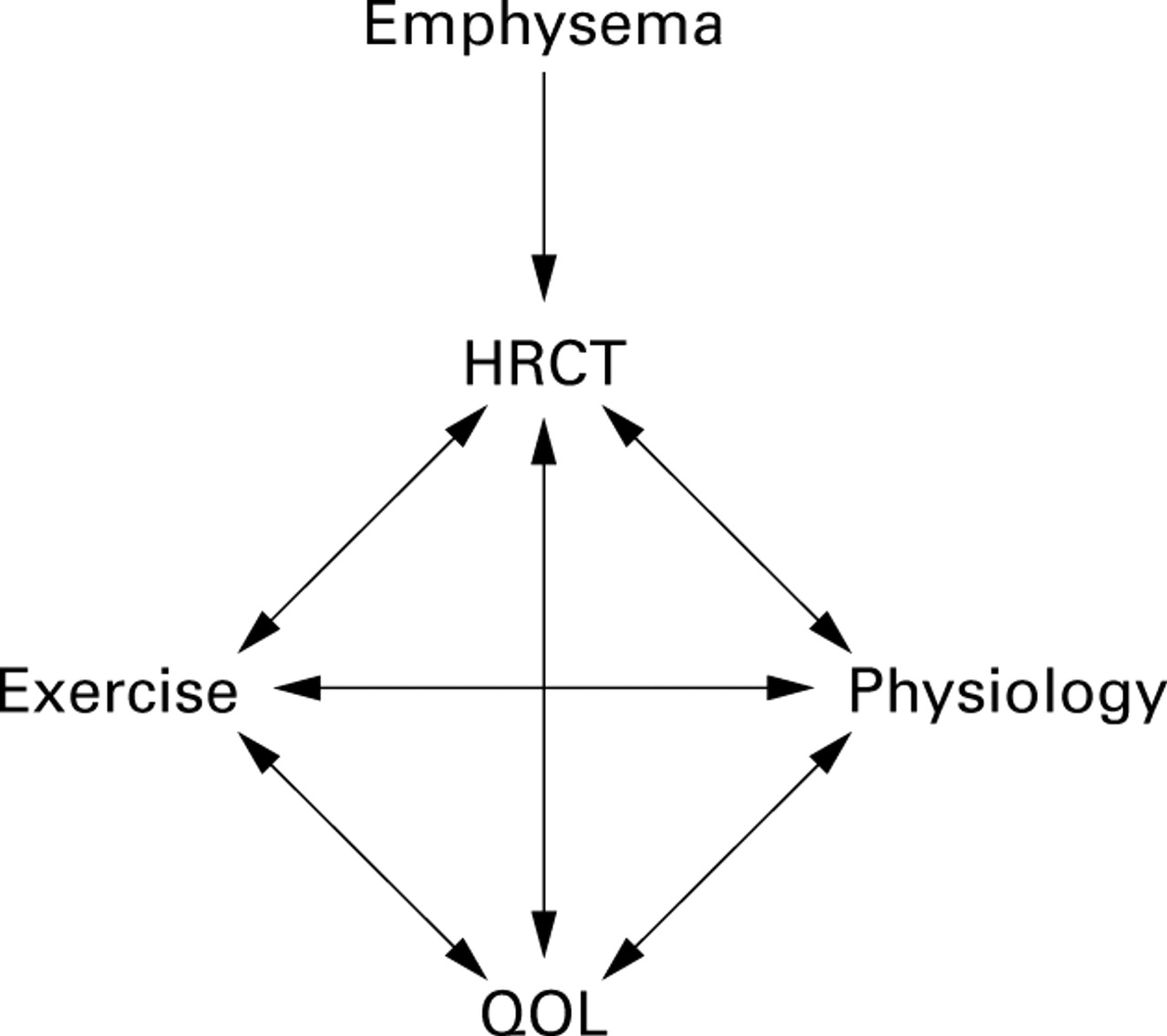
From a patient's point of view, health status is more meaningful than abnormal physiology or CT scan. There are many tools currently available to assess health status including, most notably, the St George's Respiratory Questionnaire and the more generic SF36 questionnaire. It has been shown in particular that the score derived from the St George's Respiratory Questionnaire deteriorates with time as lung disease progresses33 and this is related (although loosely) to the FEV1.34 Furthermore, the ability of patients to exercise is also reflected in their health status as well as their physiological impairment. Recent data in α1-antitrypsin deficiency have shown that both exercise and health status are related not only to the physiology but also to the degree of abnormality on the CT scan (fig 2). Although health status tools are now becoming widely accepted for patient assessment, it remains uncertain whether these will be sensitive enough to be included as primary or secondary outcome measures in clinical trials of augmentation therapy.
{kind=link}
{kind=link}
Interrelationship between tools to assess patients with α1-antitrypsin deficiency.
Is a controlled trial possible?
Research in the last few years in α1-antitrypsin deficiency has provided a much better understanding of the processes involved in the development of emphysema and its progression. Studying lung inflammation through a variety of biochemical and cellular markers provides a simple means to determine whether intervention therapy reduces airway inflammation and this can be established in a limited number of patients. Such studies would appear to be far more relevant and critical than the early work merely demonstrating an increase in lung α1-antitrypsin levels with augmentation therapy. High resolution CT scanning is relatively specific for the presence of emphysema and is objective, reproducible, and demonstrates progression indicating greater sensitivity to change than the FEV1. Furthermore, objective analysis of the CT scan relates not only to health status but also to other tests of physiological impairment which strengthens its role as a major outcome measure in any clinical trial of augmentation therapy. Such an approach would markedly reduce the number of patients required to demonstrate a positive effect and the period of time over which monitoring would be necessary, thereby strengthening the feasibility of implementing such a trial. However, in order to facilitate a controlled trial it will be necessary to identify sufficient patients who have not yet received augmentation therapy.
As indicated earlier, many countries routinely prescribe augmentation therapy to a significant proportion of patients. In 1997 interested clinicians in charge of national registries from 12 countries joined together to develop an international registry for α1-antitrypsin deficiency. In 1999 AIR (Alpha-1 International Registry) was formed with a coordinating committee, council, and formal statutes. The aims of this group are to continue research into α1-antitrypsin deficiency and its treatment and to develop a major database (which currently includes patients from Sweden, Denmark, Holland, Belgium, Austria, Germany, Spain, Italy, Switzerland, Australia/New Zealand, South Africa, Canada, Japan and the USA). The current UK registry includes over 400 patients, none of whom are receiving augmentation therapy. This, together with Sweden (more than 900 patients), Denmark (more than 900 patients), and Holland (more than 400 patients), provides an extensive resource from which patients can be recruited to suitably powered controlled clinical trials. However, this remains a valuable yet limited resource. At present several pharmaceutical companies are exploring the possibility of controlled trials using human purified or transgenic α1-antitrypsin, anti-elastase drugs that can replace α1-antitrypsin function, and anti-inflammatory therapy to circumvent the need for anti-proteinase protection of the lung. It is critical that such programmes are coordinated but, in addition, are based on sound science and state of the art drug delivery and clinical monitoring. It would be tragic if, after all this time, a poorly thought through trial based on historical data fails because of attempted expediency. Such a disaster would destroy the carefully laid groundwork of the international community and prevent the instigation of better studies.