Article Text

Statistics from Altmetric.com
Since its initial descriptions in 1946,1 the measurement of airway responsiveness with inhaled bronchoconstrictor stimuli such as methacholine or histamine has become routine practice in the diagnosis and follow up of asthmatic patients. Practically, this involves the patient inhaling increasing doses or concentrations of a stimulus until a given level of bronchoconstriction is achieved, typically a 20% fall in forced expired volume in one second (FEV1). Airway responsiveness is then expressed as the provocative dose or concentration of the stimulus required to achieve this degree of bronchoconstriction (PD20 and PC20, respectively).
Definition of terms
The term “airway responsiveness” is preferred when discussing PD20 and PC20 measurements as this is a non-specific term that encompasses the underlying mechanisms that may be responsible for differences in these measurements either between individuals or within an individual over time2. This is best illustrated in fig 1 where it can be seen that a decrease in PC20 may be due to a steeper dose-response curve (hyperreactivity) or to a shift in the curve to the left (hypersensitivity), or both. Thus, when an individual displays a decreased PC20 it is usually not known whether this is due to hyperreactivity or hypersensitivity although it is certainly one, the other, or both—all of which are covered by the term airway “hyperresponsiveness”.
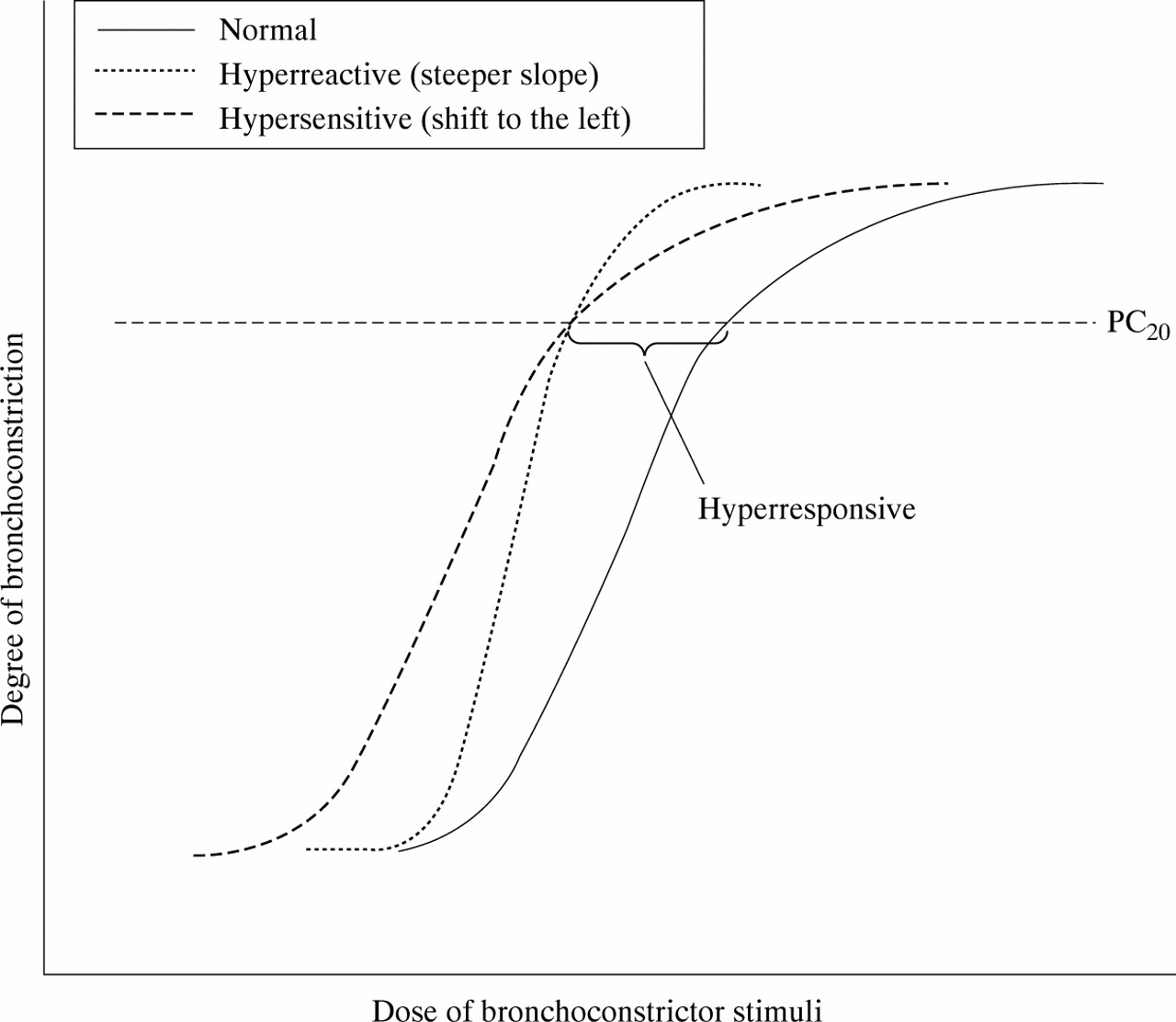
Changes in FEV1 induced by increasing doses of a bronchoconstrictor stimulus such as histamine or methacholine. Bronchial hyperresponsiveness, defined as the dose causing a 20% fall in FEV1, can be induced by airway hypersensitivity (shift to the left of the dose-response curve) or airway hyperreactivity (steeper slope of the dose-response curve). This figure illustrates how either of these two different mechanisms may result in the same degree of hyperresponsiveness (as measured by a shift in PC20).
Airway responsiveness in asthma
Airway hyperresponsiveness (AHR) is present in almost all patients with asthma, at least when they are having current symptoms.3 Furthermore, it is well established that patients with more severe asthma have more responsive airways than patients with mild disease,3-6 and that patients experiencing exacerbations of asthma—for example, during allergen exposure—develop more pronounced AHR during this period.4 ,7
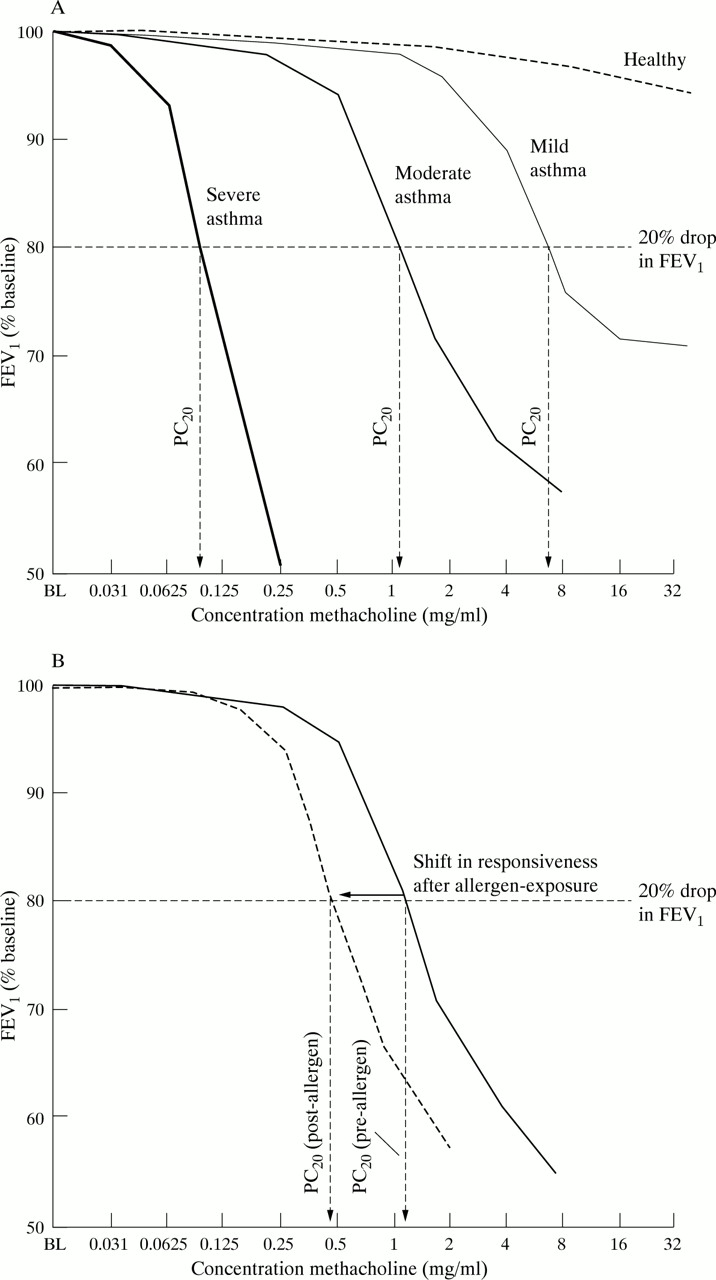
The difference in airway responsiveness between normal individuals and asthmatic patients is substantial3 being 4–8 doubling concentrations of inhaled methacholine less than in those normal subjects in whom it can be measured. In fact, no identifiable airway responsiveness can be measured in many healthy individuals as FEV1 does not fall by 20% (fig 2A). However, in individual asthmatic patients airway responsiveness is generally quite stable when the disease is stable, but can increase during exacerbations of asthma induced by allergen or other stimuli (fig 2B).

{kind=link}
{kind=link}
(A) Change in FEV1 versus baseline induced by increasing doses of a bronchoconstrictor stimulus (methacholine) in mild, moderate, and severe asthmatics compared with healthy individuals. The PC20 value is calculated by interpolating a 20% fall in FEV1 to the log linear dose-response curve for each individual. Mild asthmatic patients respond at high doses of methacholine, most often showing evidence of a plateau of the dose-response curve. By contrast, moderate and severe asthmatic subjects respond with bronchoconstriction at much lower doses and, in the most severe cases, no plateau of the dose-response curve can be found. (B) The degree of shift in bronchial hyperresponsiveness in an asthmatic individual having been exposed to allergen is on a mean level approximately 1–2 doubling doses of methacholine/histamine. This degree of shift is much smaller than the difference seen between asthmatic patients and normal individuals (A).
The aim of the present review is to discuss the association between allergen induced changes in airway responsiveness and other markers of disease severity including symptoms and inflammatory indices. Following a short discussion of the possible mechanisms responsible for airway hyperresponsiveness, we will examine changes in responsiveness associated with natural allergen exposure as well as experimental allergen challenges with both high and low doses of allergen, and how these changes in AHR correlate with changes during the clinical course of asthma. An important focus of this review will thus be to establish whether experimental increases in AHR as a result of allergen challenge are clinically relevant. Finally, we will discuss which practical issues we regard to be important when designing allergen challenge based studies and when interpreting studies already published.
Mechanisms of AHR
Many different factors have been suggested to be involved in the AHR seen in asthma. Fundamentally different inflammatory processes are thought to be important. It is believed that the increased number of eosinophils in asthmatic airways produces many of the tissue changes seen in the disease, including epithelial damage, thickening of the basement membrane, and the release of mediators with the capacity to cause bronchial smooth muscle contraction and exudation of plasma, resulting in thickening of the airway wall. Indeed, it is possible that a number of these different mechanisms interact to produce AHR, but it seems that different mechanisms are involved in causing different components of AHR. Firstly, it is likely that one separate mechanism is responsible for the underlying hyperresponsiveness in asthmatic patients, differentiating them from normal individuals. Secondly, another mechanism seems to be important for the changes in AHR seen within asthmatic subjects during the course of the disease.
MECHANISM OF THE UNDERLYING AHR
It seems plausible that the airway wall thickening seen in asthma could explain some of the differences in AHR between normal individuals and asthmatic patients. For example, it has been reported that the thickness of the airway wall from necropsy specimens is greater in subjects with fatal asthma than in those with milder disease and in non-asthmatics.8 It is not exactly clear which tissue contributes most to thickening of the airway wall in asthma, but the subepithelial thickening seen in bronchial biopsy specimens from most asthmatics9 may be involved. Furthermore, the bronchial smooth muscle may have a larger volume in asthmatic subjects, at least in patients with more severe disease. Lastly, exudation of plasma can cause oedema, and thus thickening, of the airway wall.10-13 Together these factors may, by geometric mechanisms, enhance the airway luminal resistance induced by a certain degree of airway smooth muscle shortening.14 Another feature of the asthmatic airway that correlates with the degree of AHR is loss of epithelial structure15 or the appearance of epithelial cells in bronchoalveolar lavage fluid.16 The partial loss of the epithelial barrier may allow greater amounts of bronchoconstrictor mediators to reach the smooth muscle or other cells which amplify the bronchoconstricting effect of the inhaled mediators. Alternatively, the release of bronchodilating substances from the epithelium17 could be reduced in states of epithelial damage which could enhance bronchial smooth muscle contraction induced by inhaled or endogenous bronchoconstrictor stimuli. One such epithelial bronchodilating factor is PGE2 18 ,19 but it seems that non-prostanoid factors are also involved.
MECHANISM OF CHANGES IN AHR
Bronchial wall eosinophilic inflammation is a prominent feature of asthma and it has been suggested that chronic eosinophilic inflammation contributes to the development of a persistent AHR by mechanisms of inducing airway wall thickening. However, it seems unlikely that ongoing eosinophilic inflammation by itself would be the sole cause of AHR because eliminating the inflammation by glucocorticoids only improves, but does not eliminate, AHR.20 However, fluctuations in the extent of eosinophilic inflammation may underlie the changes in AHR seen during the course of the disease. The number of eosinophils, as well as their activation as judged by surface EG2 staining in bronchial biopsy specimens and lavage fluid from asthmatic subjects, correlated inversely with AHR.21 Furthermore, different forms of allergen exposure such as challenge with a single dose of allergen,22-24 exposure to repeated low doses of allergen,25 ,26 or seasonal exposure to a pollen allergen,27 all of which increase eosinophilic airway inflammation as judged by biopsy specimens, lavage fluid or induced sputum, enhance the AHR that is already present in these individuals. Furthermore, eliminating or decreasing allergen load by allergen avoidance measures only improves, but does not eliminate, AHR.28-30
While we have proposed that there may be separate mechanisms responsible for the underlying AHR of asthma and for the fluctuations seen throughout the course of the disease, it is quite likely that this distinction is not complete. Clearly it is possible, if not probable, that the underlying mechanisms responsible for inflammatory cell recruitment and mediator release may, in the short term, be responsible for fluctuations in AHR and, in the long term, for the underlying structural changes responsible for chronic AHR.
AHR AND OTHER ASTHMA VARIABLES
AHR has been shown to increase during natural exacerbations of asthma. Increased variability of peak expiratory flow and symptoms of asthma are generally associated with histamine or methacholine AHR.31-33 Furthermore, inducing a slight worsening of asthma by withdrawing inhaled glucocorticoids34 increases mean peak expiratory flow variability from approximately 12% to 18%, which is associated with an increase in methacholine AHR of approximately one doubling dose. This experimental asthma exacerbation was also shown to be associated with increased numbers of both circulating and sputum eosinophils, suggesting a worsening of airway inflammation which may explain the increase in responsiveness.
One intrinsic problem with quantifying AHR during an exacerbation of asthma is, however, that airway calibre may be reduced per se which would increase the AHR measured by purely geometric mechanisms.32 Changes in AHR in more severe exacerbations of asthma, causing overt changes in lung function, cannot therefore be quantified accurately.
Changes in AHR during natural exposure to allergen
Natural exposure to allergen includes seasonal pollen inhalation and house dust mite inhalation. The first study to show increases in AHR in asthmatic subjects during seasonal allergen exposure was published by Altounyan in 1979.35 The degree of increase in AHR with natural allergen exposure varies between patients and probably with the dose of exposure, but in a population of asthmatics the mean changes in AHR to cholinergic stimuli are of the magnitude of 1–2 doubling doses.27 ,35-38 Based on these observations which show that certain features of asthma are worsened following allergen exposure, a number of experimental studies have been performed with experimental allergen challenges. These studies have in many ways increased our understanding of the underlying mechanisms of asthma, and contributed to the development of drugs useful in the management of asthma.
CHANGES IN AHR AFTER SINGLE ALLERGEN EXPOSURE
The first published study to demonstrate increased AHR following single dose airway allergen challenge was by Cockcroft et al.4 In this study the increased histamine AHR following allergen challenge was found to last for up to seven days and was more likely to occur in subjects who developed dual asthmatic responses (early and late bronchoconstriction) than in isolated early responders, a finding which has been supported by several subsequent studies.39-41 In a recent study involving 31 asthmatic subjects we have measured an average shift in methacholine PC20 of 1.39 doubling concentrations 24 hours after a single dose allergen challenge, producing early and late asthmatic responses reflected by at least 15% falls in FEV1.42 The increase in AHR after allergen inhalation challenge has been associated with increases in a number of markers of airway inflammation including circulating levels of eosinophils, eosinophil cationic protein (ECP), and basophils,39 ,43 bronchoalveolar lavage fluid levels of activated eosinophils and lymphocytes,22 ,23 ,44 ,45and levels of eosinophils, neutrophils and metachromatic cells in induced sputum.24 ,46 ,47
CHANGES IN AHR DURING REPEATED LOW DOSE ALLERGEN EXPOSURE
A number of studies with experimental low dose allergen exposure in mild asthmatic subjects have recently been reported.25 ,26 ,48 These studies have all been performed in patients with asthma symptoms associated only with allergen exposure. In two of the reported studies the dose of allergen was individualised on the basis of the patient’s sensitivity to allergen and a dose of 5–25% of the dose causing an early and late asthmatic response was generally used (greater than 15% decrease in FEV1), given on 4–7 consecutive days, whereas a randomly chosen low dose was selected for the older study.48
These studies suggest that methacholine AHR increases after repeated low dose allergen exposure. It had been argued that this change in AHR was unaccompanied by increased inflammation.48 However, in two more recent studies increased plasma levels of ECP and airway sputum eosinophilia were associated with the increased airway responsiveness.25 ,26 In these studies the degree of increase in AHR depended on the time of measurement. A maximal change was observed eight hours after the third low dose allergen exposure and amounted to approximately two doubling doses compared with repeated placebo/vehicle.26 This degree of change in AHR was maintained for at least another two days of low dose allergen exposure.26 The increase in AHR induced by low dose allergen exposure seems to recover to basal levels quite rapidly and 24–30 hours after the last dose the observed degree in change was only approximately one doubling dose,25 ,26 and 72 hours later the induced changes in AHR were no longer statistically significant.26
In these two recent studies it was clearly shown that repeated exposure to low dose allergen causes both symptoms of asthma25 ,26and increased variability in lung function,25 suggesting that experimentally increasing AHR by approximately 1.5–2 doubling doses is associated with signs of clinical worsening of asthma.
CLINICAL RELEVANCE OF INDUCED CHANGES IN AHR
The reviewed studies suggest that exposure to a single allergen, to repeated low dose allergen, to natural allergen and spontaneously occurring exacerbations of asthma all enhance methacholine or histamine AHR by, on average, 1–2 doubling doses from a stable baseline. These changes in AHR are likely to be clinically significant since they occur in addition to the persisting AHR that is present in all asthmatic subjects and are associated with increases in variability of lung function and symptoms of asthma.
It has been suggested that indirect challenges such as isocapnic dry air hyperventilation, exercise, or inhalation of adenosine monophosphate (AMP) would illustrate the inflammatory state of the airways better than methacholine or histamine challenges.49-51 It is therefore possible that different challenge methods may be used to understand different aspects of the underlying mechanisms of the change in AHR induced by allergen exposure.
MEASURING ALLERGEN INDUCED AHR
The proven association between allergen induced AHR and allergen induced inflammation supports the many studies where the efficacy of several classes of anti-asthma drugs has been tested by their ability to prevent increases in AHR following allergen inhalation challenge.52-63 To date all investigations of this type have been based on the single allergen challenge protocol. Typically, in these studies sample sizes ranging from eight to 20 subjects are used in a repeated measures crossover design to test for differences between treated and placebo responses.
We have recently performed a series of power calculations to determine whether these sample sizes are adequate for investigating treatment effects on allergen induced airway responses. When doing this we have assumed that a treatment induced reduction of 50% in any allergen induced deterioration in asthma is clinically significant (including early and late asthmatic responses and changes in airway responsiveness). Based on this assumption we have previously shown that sample sizes of approximately 10 subjects are sufficient to demonstrate clinically significant effects (50% attenuation) on the magnitude of both the early and late asthmatic responses.64 More recently, however, we have evaluated what would be the appropriate sample sizes to determine the efficacy of various treatments on allergen induced increases in airway responsiveness.42In this study 31 subjects with dual asthmatic responses following airway allergen challenge showed a mean fall in methacholine PC20of 1.29 doubling concentrations. In the same study we calculated the average standard deviation of the difference between placebo and actively treated PC20 shifts in several published trials and found it to be 0.96 doubling concentrations. Based on these observations we calculated the number of subjects required to demonstrate clinically significant effects on allergen induced AHR (50% attenuation) in repeated measures trials. Assuming a desired power level of 90% we found that 24 subjects would be required to demonstrate, with a 90% power, that a treatment can attenuate allergen induced AHR by 50%. Obviously fewer subjects would be required when studying more effective treatments, but it is clear that sample sizes currently employed in studies of this type are inadequate, resulting in low power to show inhibitory effects on allergen induced AHR.
While there have been few studies to date quantifying the changes in AHR following multiple low dose allergen challenge, it is clearly important that appropriate sample size requirements should be established before this protocol becomes widely used to test the efficacy of anti-asthma drugs. Based on our initial study with eight subjects we have observed an average increase shift in PC20of 1.72 doubling concentrations with a standard deviation of 0.32 doubling concentrations in the difference between the allergen and diluent conditions. Assuming that this standard deviation will be close to that measured when comparing allergen responses following active treatment and placebo, drugs resulting in a 50% attenuation of the allergen induced shift in PC20 could be detected with fewer than five subjects (90% power). This higher power of the repeated low dose challenge compared with the more widely used single high dose challenge clearly needs to be further investigated before studies are performed with such small sample sizes.
Conclusion
Inhaled allergens initiate processes that increase airway inflammation and enhance AHR in asthmatic subjects. Thus, studies using inhaled allergen challenges have provided insight into how changes in AHR are regulated by induced inflammatory processes. These changes in AHR (1–2 doubling doses) have been shown to be of much smaller magnitude than those seen when asthmatics with stable AHR are compared with normal subjects in whom AHR can be measured (differences of 4–8 doubling doses). These allergen induced changes are, however, important as they are similar to changes occurring in asthmatic subjects who already have AHR that are associated with worsening asthma control. It is likely that the mechanisms responsible for the changes in AHR following experimental allergen exposure are similar to those producing transient worsening of control in asthmatics. Nevertheless, the mechanisms of the transient allergen induced AHR are not likely to explain the underlying mechanisms of the persistent AHR in asthmatic patients when compared with normal individuals.
Acknowledgments
JL is financed by the Vårdal Foundation and the Swedish Medical Research Council