Article Text

Statistics from Altmetric.com
ADAM 33, the latest of the ADAM proteins to be described, has been identified as a major susceptibility gene in asthma linked to bronchial hyperresponsiveness. It provides an important breakthrough in our understanding of this complex disorder and its variable clinical and physiological presentations.
Asthma is a disorder of the conducting airways in which Th2 mediated inflammation interacts with structural changes to cause variable airflow obstruction. Fundamental to disordered function is the concept of bronchial hyperresponsiveness (BHR) in which the airways constrict too much and too easily. In chronic severe asthma the inflammation and structural changes both become more intense1 and are paralleled by an increase in BHR that is only partially or non-responsive to treatment with corticosteroids.2 Explanations for BHR include mucosal and adventitial swelling causing a disproportionate reduction in airway calibre for a given degree of airways smooth muscle (ASM) shortening,3 excessive ASM shortening,4 an increase in ASM mass causing greater force generation,5 and an excessive velocity of contraction linked to altered crossbridge cycling.6 Morphometric studies have shown a graded increase in ASM mass in proportion to disease severity, and computer modelling has revealed that this and altered contractility are the most plausible explanations for BHR.5,7
IDENTIFICATION OF A NEW ASTHMA RELATED GENE
Asthma has a high heritability of up to 79%,8 involving a few genes with moderate effects rather than many genes with small effects.9 We have recently reported the first novel asthma related gene identified by positional cloning.10 A genome wide screen was undertaken using phenotypic data and DNA from 362 families in Wessex and 98 in the USA with at least two siblings with asthma. Using 401 microsatellite markers at a density of 9 cM and multipoint linkage analysis, suggestive evidence for linkage (maximum lod score (MLS) 2.24) was found on the short arm of chromosome 20 at 9.99 cM. The addition of 13 more markers at 1–2 cM intervals increased the MLS to 2.94 at D20S482 (12.1 cM) which further increased to 3.93 when BHR was included in the definition of asthma (despite halving the sample size), thereby exceeding the threshold for genome wide significance.11 In contrast, when asthma was conditioned for serum total IgE and allergen specific IgE as measures of the allergic component of asthma, the MLS fell to 2.3 at 11.6 cM and 1.87 at 12.1 cM. This indicated that the chromosomal region under the peak of linkage contained genes more closely linked to altered airway function than to measures of atopy. Confirmation for linkage on chromosome 20 has come from separate analysis of the UK and US families,10 and from two separate genome wide studies in other UK and US outbred populations.12,13
Physical mapping of the region under the peak of linkage on 20p13 and direct cDNA selection led to the identification of 40 genes.10 Single strand conformational polymorphism (SSCP) analysis and direct sequencing was used to identify single nucleotide polymorphisms (SNPs) used for case-control association studies involving 130 affected individuals who contributed to the linkage signal on chromosome 20 and 217 controls with no personal or family history of either asthma or allergy—that is, “hypernormals”. In 23 genes spanning the 90% confidence interval on each side of D20S482, 135 SNPs were typed of which 25 were localised to a cluster of five genes showing significant association with both asthma and BHR. Fourteen of these lay within a single gene identified as ADAM 33, a novel member of the ADAM (A Disintegrin And Metalloproteinase) family (achieving significance of p=0.005–0.05). Both in the combined populations and in the UK and US samples when analysed separately, additional SNP typing strengthened the location of the signal to ADAM 33 and this was further confirmed both by haplotype analysis (5–7 SNP combinations at p=0.000003–0.005) and transmission disequilibrium testing (TDT) (8 SNP combinations at p<0.005). The association of SNPs in ADAM 33 with asthma has recently been confirmed in three different ethnic populations in the USA, one in Holland,14 and in separate case-control and family studies in Germany (M Wijst, personal communication).
CHARACTERISTICS OF THE ADAM GENE FAMILY
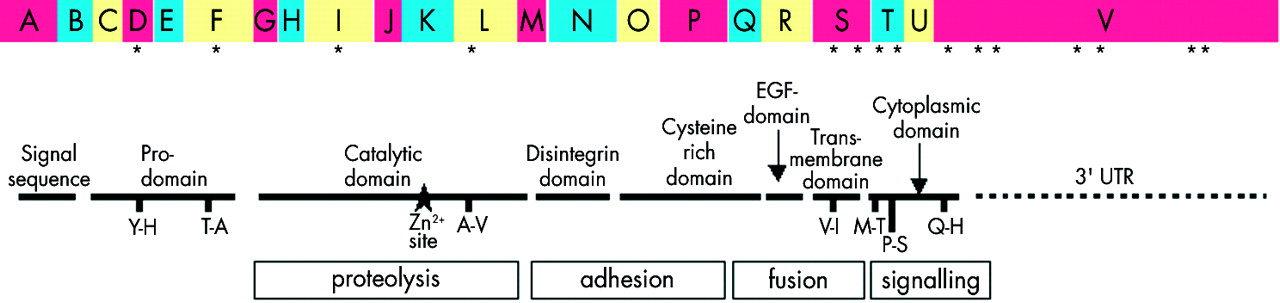
ADAM 33 is the most recently reported member of the ADAM gene family of Zn++ dependent matrix metalloproteases (MMPs). ADAMs have a complex organisation involving eight domains, the first six encoding signal sequence and pro-, catalytic, disintegrin, cysteine rich and EGF domains15 which are anchored at the cell surface or Golgi apparatus by a transmembrane domain, followed by a cytoplasmic domain with intracellular signalling specific sequences (fig 1). ADAM 33 belongs to the ADAM 12, 15 and 19 subfamilies, all of which possess proteolytic activity.15,16 Northern blot analysis identified two transcripts of ADAM 33 of 5.0 and 3.5 kb, but only the latter has been found in cytoplasmic RNA.10 Seventeen cDNA libraries have been screened resulting in the identification of a 3.5 kb clone containing the entire open reading frame comprising 22 exons with the canonical sequence (CT/AG) present at each splice junction. The potential for alternatively spliced transcripts is also indicated by the identification of variants lacking exons, or parts of exons. ADAM 33 is preferentially expressed in smooth muscle, myofibroblasts and fibroblasts, but not in epithelial cells, T cells or inflammatory leukocytes.10,17 The significance of ADAM 33 for asthma is strengthened by the existence of a syntenic region on mouse chromosome 2 at 74 cM that has been linked to BHR18 overlying an orthologue of ADAM 33 that exhibits approximately 70% homology with its human counterpart.17,19
{kind=link}
The exon structure and domain organisation of full length ADAM 33. The location of disease related SNPs in the coding and 3′ untranslated regions are indicated by an asterisk and those SNPs leading to an amino acid change are also shown together with the likely functions of each domain. Reproduced with permission of Elsevier from Davies et al.22
The selective expression of ADAM 33 in mesenchymal cells strongly suggests that alterations in its activity may underlie abnormalities in the function of ASM cells and fibroblasts linked to BHR in asthma. These components of the epithelial-mesenchymal trophic unit (EMTU)20 share a common fibroblastic progenitor, the mature phenotype of which is directed by growth factors, including TGF-β,21,22 whose release from bronchial epithelial cells is increased in response to epithelial damage and airway inflammation.23,24 Asthmatic (myo) fibroblast cells are unusual in that they have the capacity to proliferate in the absence of exogenous growth factors,21,25 paralleling the behaviour of asthmatic ASM cells in vitro.26 This shared trait is consistent with the occurrence of a common stem cell population whose numbers are increased in asthma due to an inherent capacity for enhanced growth and/or survival.
Members of the ADAM family are proteins with diverse functions that reflect the complex domain structure of these molecules (fig 1).15 While certain functions can be attributed to an individual domain—for example, ectodomain shedding to the metalloproteinase domain27 and cell adhesion to the disintegrin domain28—it is likely that the other domains play important regulatory roles in these functions by conferring specificity and selectivity. ADAM proteins are anchored in the trans-Golgi network or plasma membrane but, in some cases, secreted splice variants have been identified. In the case of ADAM 12, an evolutionarily close relative of ADAM 33, ectopic expression of the secreted form of the molecule (ADAM 12S) in rhabdomyosarcoma cells results in growth of tumour xenografts which are infiltrated with large numbers of host derived smooth muscle cells.29 Although we have identified several alternatively spliced forms of ADAM 33 in lung derived cDNA, it is not yet known whether a secreted protein variant is produced by airway cells.
The importance of genetic variation in ADAM proteins has recently been highlighted by mutations in ADAMTS13 (ADAM with thrombospondin type 1 motif) that underlie thrombotic thrombocytopenic purpura,30 while decreased levels of ADAMTS2 impair collagen type 1 processing leading to fragile skin in Ehlers-Danlos syndrome.31
POSSIBLE ROLE OF ADAM 33 IN ASTHMA AND BHR
Although ADAM 33 is a highly polymorphic gene containing over 55 SNPs, genetic analyses have identified the 3′ portion of the gene that encodes the transmembrane and cytoplasmic domains and 3′UTR as likely to be the key region linked to asthma and BHR.10 In the UK population association studies revealed that six SNPs in the ADAM 33 gene were significant at p<0.03, including two (S1 and S2) in the exon encoding the transmembrane domain (p=0.03 and 0.004, respectively) and another (ST+4) which lies in the intron separating the transmembrane and cytoplasmic domains (p=0.02, fig 1). When ST2 and ST+4 were analysed as a haplotype or with SNPs in the intron preceding the V exon, the level of significance greatly increased (p=0.000003–0.0005). Further confirmation that the transmembrane and intracellular domains are key sites for controlling altered functions of ADAM 33 comes from TDT analyses in the UK population. In this case the exonic S1 SNP, which leads to an amino acid substitution (V→1 (one letter code)), showed a significant association with asthma (p=0.0043) and BHR (p=0.0066). Additional haplotype analysis increased the significance of the association of this SNP with asthma and BHR when taken in conjunction with the T1, an SNP which causes an amino acid substitution (M→T) in the cytoplasmic domain, and with SNPs in the V exon encoding the end of the cytoplasmic domain and the 3′UTR (p=0.0001). Considering that the S1 and T1 SNPs occur in close proximity in the transmembrane and cytoplasmic domains of ADAM 33 and flank the intronic ST+4 SNP (fig 1), polymorphisms in this region together with others in the 3′UTR may alter the activity of ADAM 33, conferring susceptibility to BHR and asthma. However, without further genetic and functional studies it is difficult to attribute primacy to any individual SNP because of the extent of linkage disequilibrium that exists in this region.
The cytoplasmic domains (CTD) of ADAM proteins are typically rich in proline and have important regulatory functions through their interaction with intracellular signalling proteins via SH2 and/or SH3 domains. Furthermore, the presence of alternatively spliced cytoplasmic variants of ADAM 22 have been shown to exhibit cell selective expression. Yeast two hybrid, western blotting, and pull down assays have identified several proteins that interact with the ADAMs, including src family kinases, endophilin, α-actinin, PACSIN, and protein kinase C (PKC).27,32–35 Together with the transmembrane domain, the CTD mediates interactions with the cytoskeleton and is involved in intracellular trafficking of the protein.36 For example, phosphorylation of ADAM 9 by PKC causes trafficking of the protein from the Golgi to the plasma membrane where it cleaves heparin binding EGF-like growth factor (HB-EGF) from its membrane bound precursor.27 In the case of ADAM 33, the intracellular domain is relatively short by comparison with its nearest homologues but it is very rich in prolines and has a putative SH3 binding site (PsWPLDP) in which the T2 SNP is located and may affect function. There is also a casein kinase F/II phosphorylation site (SHEPSSHP) and a MAPK consensus sequence which could be important regulators of ADAM 33 function. Although the T1 SNP causes an M→T substitution, no new recognisable phosphorylation site is generated.
ADAM 33 is amply provided with a number of other mechanisms whereby its disordered function could translate into BHR although, at this time, it is not clear whether changes in ADAM 33 linked to asthma relate to a gain or loss in function.37,38 In many ADAMs the proteolytic site has the capacity to release cytokines and growth factors from their precursors. For example, ADAM17 (TNFα converting enzyme (TACE)) is responsible for generating both TNFα and TGFα,39 while ADAMs 9 and 12 cleave HB-EGF from its precursor.27,28 In a mouse model of hypertension induced cardiac hypertrophy a pivotal role for myocardial ADAM 12 has been shown through its capacity to generate HB-EGF.40 Loss of function may lead to impaired processing of cytokines and growth factors. Reduced processing of myostatin, a member of the TGF-β/BMP superfamily, is associated with differentiation into myotubes.41 Several of the ADAM family members are involved in cell-cell and cell-matrix interactions involving members of the integrin family,42 although binding to syndecans,43 members of the tetrapsanin family, and unknown proteins has also be shown. Binding of fibroblasts to a soluble ADAM 9:Fc fusion protein via α6β1 causes a very large increase in cell motility, which suggests that the ADAM could act as a locomotor adhesion receptor.44 In contrast, overexpression of ADAM 15 decreases cell migration by increasing cell-cell contacts without affecting initial cell adhesion.45
In conjunction with the cysteine rich domain, a novel secreted form of ADAM 12 provokes myoblast fusion in vivo by binding to the muscle specific actin binding protein, α-actinin-2.29 If the equivalent domain in ADAM 33 has similar properties, it could be implicated in converting ASM cells from acting as multiple units to a single contractile unit,46 a property relevant to the pathogenesis of BHR.
CONCLUSIONS
The identification of ADAM 33 as a major susceptibility gene in asthma linked to BHR is an important breakthrough in our understanding of this complex disorder and its variable clinical and physiological presentations.47 ADAM 33 is the latest of the ADAM proteins to be described,17,19 with its implication in the pathogenesis of asthma being discovered in the same year. Identifying a major new candidate gene for asthma creates an imperative to determine its function and how disordered function translates into disease. The work ahead is likely to provide important insights into the origins and progression of asthma, especially since expression of ADAM 33 has now been shown in fetal lung tissue. It is not infrequently stated that searching the whole genome for susceptibility genes in complex diseases such as asthma is a fruitless “fishing expedition”. However, as recently pointed out by Ahmadi and Goldstein,47 a sequential approach to identifying novel genes in complex disorders by combining linkage and association approaches can achieve success without having to resort to expensive and time consuming genome-wide association mapping. To achieve this in any complex disease it is becoming increasingly appreciated that it is essential to define the disease phenotype accurately. ADAM 33 can now be added to calpain 10 (type 2 diabetes)48 and NOD 2 (Crohn’s disease)49 as an entirely novel molecule linked to the pathogenesis of a complex human disease. The future challenge will be to determine the function(s) of ADAM 33 and the abnormalities that may occur to account for its association with asthma and BHR.
ADAM 33, the latest of the ADAM proteins to be described, has been identified as a major susceptibility gene in asthma linked to bronchial hyperresponsiveness. It provides an important breakthrough in our understanding of this complex disorder and its variable clinical and physiological presentations.