Article Text

Statistics from Altmetric.com
Although much has been learned about the mechanisms leading to acute lung injury, mortality—which is mainly related to sepsis or associated non-pulmonary organ dysfunction1—remains high (around 50%) in patients with adult respiratory distress syndrome (ARDS).2-4 Many new therapeutic approaches aiming to control the inflammatory response accompanying ARDS have been evaluated.5 However, these treatments have had no impact on the mortality stemming from the disease.5 The lack of success with these new interventions is probably multifactorial.6 One possible explanation is that the appropriate patient population had not been enrolled for study.7 In this regard, it is also probably unrealistic to hope that a single treatment will modify the evolution of all ARDS patients who represent a heterogeneous population with very different severities of lung injury. Thus, it is unlikely that the efficacious treatment of patients with mild lung injury will be as efficient in patients with severe lung injury.
Most of the treatments tested recently were targeted to control the inflammatory response.5 Although the development of lung injury is mainly dependent on aggression of endothelial cells by inflammatory cells,8 its severity and recovery also depend on epithelial cell function.9 In fact, the predominant pathological finding in acute lung injury is diffuse alveolar epithelial damage.10 ,11 Furthermore, physiologically it has been shown that the structure and function of the alveolar epithelium are important determinants of lung injury.12Finally, the alveolar epithelium is also the site of alveolar fluid reabsorption and plays a major role in the development of lung fibrosis associated with ARDS.13-15 Treatments aimed at improving epithelial function might therefore become one of the key elements to accelerate recovery and decrease the mortality of patients with ARDS.
In this review we will emphasise the importance of modulating two of the many functions of the alveolar epithelium which we consider to be strategically necessary for quicker recovery from ARDS. After reviewing the characteristics of ARDS lesions, we will determine how we can stimulate the alveolar epithelium to increase oedema clearance and hasten epithelial repair.
The alveolar epithelium as a target of lung injury
Pathological observations of acute lung injury derive mainly from the analysis of necropsy material from patients who died of ARDS. By definition, these studies were done on subjects with severe lung injury and might not be representative of the pathological lesions present in the overall population of patients with acute lung injury. However, from these studies we can identify the different stages of the pathological process.
In the acute stage, corresponding to the first few days (<7) after the onset of injury, we mainly observe an exudative process characterised by extensive epithelial and endothelial barrier damage, resulting in the flooding of alveolar air spaces with proteinaceous liquid, inflammatory cells, and fibrin.10 ,11 Hyaline membranes are also seen in the alveolar spaces, especially at the end of this stage.10 ,11 This pathological stage is often categorised as diffuse alveolar damage.10 ,11 ,16 As the name suggests, the most conspicuous structure which is damaged is the alveolar epithelium—more precisely, the alveolar type I cells—which seem more sensitive to injury than alveolar type II cells.17 Endothelial cell injury is subtle and seems relatively minor to explain the rapid and continuous escape of protein-rich fluid from the microcirculation.10 Why there is such a disparate appearance of lesions between epithelial and endothelial cells is still a mystery, but one hypothesis is that the repair phase of endothelial cells is much more rapid than for epithelial cells. This also suggests that the alveolar epithelium may need some help to hasten the recovery phase from diffuse alveolar damage.
The second more chronic stage of acute lung injury (>7 days) is marked by its variability. In certain patients there is, in fact, rapid reabsorption of alveolar oedema fluid and repair of the injured region of the alveolar epithelium, followed by clinical recovery from respiratory failure. However, in a number of patients alveolar oedema persists and we notice the organisation of hyaline membranes and gradual appearance of intra-alveolar fibrosis.10 ,11 The development of this intra-alveolar and interstitial fibrosis distorts the normal architecture of the lung and prevents the recovery of patients since such an abnormal structure is incapable of performing adequate gas exchange.
Thus, one of the key factors to recovery from ARDS is the ability to clear oedema fluid and reconstitute a normal alveolar structure. To be able to stimulate these processes we have then to understand the basic physiological mechanisms behind these two functions.
Lung alveolar epithelial cells: pluripotent cells of the distal air spaces?
More than 20 years ago Mason et alpictured alveolar type II cells as crenated towers defending the alveolus.18 At this time virtually nothing was known about the functional abilities of these cells, and their in vitro story had just begun. Almost a quarter of a century later it can be stated that alveolar type II cells are not innocent bystanders but pluripotent cells involved, not only in surfactant secretion, but also in ion transport and epithelial repair. Although much is known about the surfactant secreting function of these cells, their role in ion transport and epithelial repair is not as well understood.
HOW DOES OEDEMA LIQUID LEAVE THE AIR SPACES?
The alveolar epithelium is a key element in the process of oedema clearance (fig 1). In fact, the alveolar epithelial barrier is not only a tight epithelium but it is also actively involved in the transport of ions and solutes.13 Three different experimental approaches have led to demonstrations of active ion transport in the alveolar region of the lung.

Schematic representation of the major pathways for ion and water transport involved in oedema clearance in the normal lung. The mechanisms that could explain how β adrenergic agonists can increase the activity or expression of Na+ transport proteins are also illustrated.
The first experimental strategy involved a model of alveolar flooding where autologous serum or an isotonic fluid containing trace amounts of albumin was instilled in the air spaces of the lung. Fluid clearance was then measured by determining the amount of excess water in the experimental lung compared with a control lung using a modified gravimetric method19 or by quantifying over time changes in the concentration of a non-permeable molecule (such as albumin) instilled into the lung with the fluid.20 With such an experimental approach it was established that liquid clearance was faster than protein clearance, so that the solution remaining in the air spaces became more concentrated as the liquid was removed.19 ,21 Since water clearance in the face of rising osmotic pressure could not be explained by changes in hydrostatic or osmotic forces across the alveolar epithelium, it was proposed that active transport of ions could be responsible (fig1).19 ,21 This liquid clearance with rising protein concentration has been seen in the lungs of many species such as sheep,19 dogs,22 rats,23 and rabbits24 as well as humans.25 Although initially it was postulated that liquid reabsorption was occurring in the alveolar region only because it represented the largest surface area for reabsorption, recent experimental data have delivered new evidence to support such an hypothesis. Using alveolar micropuncture, Berthiaume et al 22 were the first to show that the increase in protein concentration is identical in a sample of airway fluid taken from the distal airways and the alveolar micropuncture sample. This clearly suggests that liquid absorption occurs in the alveolar region. More recently Ballardet al 26 used fluorocarbon to trap the instilled liquid in the very distal portion of the lung, mainly the alveolar region. With this model they could show that there is significant liquid absorption, again suggesting that liquid absorption occurs in very distal airways, probably in the alveolar region.
Although these data indicate active ion transport in the distal airways, further support of this hypothesis has come from experiments where lung liquid clearance was studied in the presence of inhibitors of ion transport. Ouabain, a Na+-K+-ATPase inhibitor, significantly decreased Na+transport27 ,28 and fluid absorption25 ,28 ,29 in isolated lungs, suggesting that suppression of transepithelial ion transport is important in the process (fig 1). The presence of an active ion transport mechanism was further confirmed by Serikov et al 30 who studied the effect of temperature on alveolar lung liquid clearance in isolated perfused goat lungs. In four hours they saw a 26% increase in protein concentration over baseline, similar to the result observed in sheep.20 However, when the temperature of the perfusate was decreased to 18°C there was no change in alveolar protein concentration, indicating that alveolar liquid clearance had been inhibited. These results confirmed that liquid clearance is not dependent so much on passive diffusion as it is on an active transport process.
Much experimental evidence suggests that active Na+transport is the dominant ion transport mechanism involved in this process. Amiloride, a drug well known to interfere with Na+transport,31 decreases lung liquid clearance by 40% to 75% in different species (fig 1).23-25 ,28 ,32 The potential role of Na+ in this process is also confirmed by the fact that, not only is liquid reabsorption inhibited by amiloride, but unidirectional Na+ movement from alveoli to the circulation is also suppressed by amiloride27 ,28 and ouabain.27 ,28 Finally, the relationship between lung liquid clearance and Na+ transport is further established by the demonstration that, when Na+ is replaced by choline in the alveolar instillate, there is complete inhibition of fluid reabsorption.26 ,33
The cellular mechanism responsible for this vectorial transport of Na+ from alveoli to the interstitium has been better defined recently. Firstly, the alveolar type II cell is believed to be the principal cell involved in this process since it has a greater density of Na+-K+-ATPase than the alveolar type I cell34 and it has been shown to participate actively in Na+ transport in vitro.31 Na+enters the cell by the amiloride sensitive Na+ channel (ENaC) or by other cationic channels35 located at the apical surface and is extruded by Na+-K+-ATPase located at the basolateral surface (fig 1).36 Recent experimental data have allowed us to define better the structure and function of the major system involved in this transepithelial transport.
The amiloride sensitive Na+ channel is the first constituent of the Na+ transport system. It has been cloned recently37-41 and consists of three subunits, α, β and γ ENaC, which are able to reconstitute a functional channel.38 In situ hybridisation and immunohistochemical staining have shown that the ENaC subunits are expressed along the epithelium of the respiratory system42-45 and are detected in alveolar type II cells.44 ,45 The physiological role of αENaC in the lung has been demonstrated in a mouse model where the αENaC gene was deleted by targeting a transgene by homologous recombination.46 Unable to clear liquid from their lungs, these mice die shortly after birth.46
ENaC activity and expression are regulated by a complex control system. The second messenger cAMP is thought to increase the activity of the channel (fig 1).47 Although the augmented activity of the channel is assumed to result from protein kinase A (PKA) activation and a specific phosphorylation event, it is still unclear if this involves direct phosphorylation of the channel or the phosphorylation of other proteins associated with the channel.48 The second hypothesis is more likely since PKA could increase channel activity in lipid bilayers only in the presence of actin,49 and the amino acid sequences of the three subunits contain no conserved intracellular PKA sites.48 Other physiological mechanisms have been shown to be involved in the modulation of ENaC. Recent experimental data suggest that channel activity is also dependent on channel stability at the plasma membrane, a process regulated by ubiquitination.50 ENaC function is then dependent not only on direct modulation of channel activity but also on channel degradation or stability in the membrane. The increase in Na+ transport could also be associated with an increase in channel gene expression (fig 1). Around birth there is a transient rise in ENaC expression40 ,41 ,51 at a time when liquid reabsorption in the lung is enhanced. The two major hormones that are thought to modulate the ENaC expression in the lung are catecholamines52 and steroids.51 ,53
Besides ENaC, a multitude of other cation channels that could be involved in Na+ transport have been identified in alveolar type II cells.35 One of these channels, the non-selective cation channel, has been identified on both fetal54 and adult alveolar type II cells.55 The activity of this channel can be stimulated by β adrenergic agonists56 and can be inhibited by amiloride,54 ,55 suggesting that it could be involved in Na+ transport across the alveolar epithelium. The pathways involved in channel activation by β adrenergic agonists or cAMP are still under investigation but could involve changes in the intracellular chloride concentration and Ca2+ sensitivity of the channel.56 ,57
Na+-K+-ATPase, which consists of two subunits, is another major component of the transepithelial Na+ transport system (fig 1). The α subunit is the catalytic component of the complex and is involved in Na+extrusion, K+ intrusion and ATPase activity.58The β subunit is a highly glycosylated protein whose role is not well understood but seems to be an important regulatory component of the sodium pump.58 α1 and β1subunits have been detected in the lungs.59 ,60Na+-K+-ATPase inhibition with ouabain has been shown to greatly reduce solute transport in alveoli28 and short circuit the current of alveolar type II cells.61Changes in intracellular Na+ concentration and various hormones such as mineralocorticoids, adrenergic agonists, and thyroid hormones have been found to modulate Na+-K+-ATPase activity.58 ,62Although these responses are thought to be mediated by direct protein phosphorylation by either PKA or protein kinase C (PKC), other possible mechanisms of regulation have been postulated (fig1).58 ,62 Few studies have examined the regulation of expression in normal alveolar epithelial cells. In one investigation it was shown that β adrenergic agonists increase Na+-K+-ATPase activity in alveolar type II cells.63 As with ENaC, heightened Na+-K+-ATPase expression could also be an important tactic to augment transepithelial Na+ transport (fig 1). It is well known that Na+-K+-ATPase expression is increased around birth.40 ,60 ,64 Many major hormonal systems such as the thyroid, mineralocorticoid, and glucocorticoid systems58 ,62 have been reported to modulate Na+-K+-ATPase expression. In the lung the α subunit does not seem to be modulated by corticosteroids53 but the β subunit is.65The β adrenergic system is also a potent stimulant of Na+-K+-ATPase expression in alveolar epithelial cells (fig 1).52
Until recently it was generally accepted that liquid movement across the alveolar epithelium was an intercellular process secondary to the osmotic gradient generated by transepithelial Na+ transport across the alveolar epithelium. However, several laboratories have now demonstrated the presence of specialised water transporting proteins in the lung, which could mean that water movement occurs not only through paracellular pathways but also through a transcellular route (fig1).66 Although four different members of the family of aquaporins are expressed in lung tissue, aquaporin 5 is the predominant form expressed in alveolar type I cells.67 Since this cell type has one of the highest cellular permeability coefficients for water, it could be postulated that some water movement across the alveolar epithelium is across type I cells.68 In fact, experiments performed on the isolated perfused lung have confirmed that there is significant water movement across aquaporins in the lung.66 ,69 Although there is strong evidence to suggest that water channels are physiologically relevant proteins in the lung, we know very little about their regulation. What is clear is that their expression is changed around birth70 and that modulation of the phenotype of cultured type II cells by keratinocyte growth factor (KGF) can also alter the expression of aquaporin 5.67 However, much more information will be needed to determine the possible role of these proteins in the pathophysiology of pulmonary oedema.
WHAT DO WE KNOW ABOUT OEDEMA CLEARANCE IN PATHOLOGICAL STATES?
Although there is a substantial amount of evidence to support the concept that clearance of liquid from the lung is mediated by active Na+ transport, and several pathways could be stimulated to enhance its efficiency in the normal lung, we have to wonder if this system is functional in pathological conditions. Recently, a growing number of studies have shown that the process could be functional in a number of pathological states. Some investigations have evaluated the impact of lung injury on the integrity and function of the alveolar epithelium. Interestingly, the results indicate that alveolar and distal airway epithelia are remarkably resistant to injury, particularly in comparison with the adjacent lung endothelium.36 Even when mild to moderate alveolar epithelial injury occurs, the capacity of the alveolar epithelium to transport salt and water is often preserved. In addition, several mechanisms may result in upregulation of the fluid transport capacity of the distal pulmonary epithelium, even after moderate lung injury.36 One of the models used is the sepsis model.71 ,72 In sheep, intravenous E coli endotoxin produced a marked increase in vascular permeability but had no effect on epithelial permeability or on alveolar liquid clearance.71 In the septic rats, even though endothelial injury and mild interstitial pulmonary oedema occurred after intravenous administration of endotoxin, alveolar epithelial fluid transport was augmented by 32%.72 This enhanced liquid clearance was inhibited by instillation of amiloride (10–4 M) or propranolol (10–4 M) into the distal air spaces, demonstrating that increased clearance depended on β agonist stimulation of alveolar epithelial sodium transport. Since the septic shock induced in the rats was associated with a marked rise in plasma adrenaline (epinephrine) levels, it was concluded that endogenous release of catecholamines could possibly explain the enhanced liquid clearance observed. When more severe septic shock was produced in sheep, the alveolar epithelial barrier was resistant to injury in the majority of animals with oedema being confined to the pulmonary interstitium.73 In some sheep, however, more severe systemic and pulmonary endothelial injuries were associated with alveolar flooding, a striking rise in epithelial permeability to protein, and the inability to transport fluid from the air spaces of the lung.73 The inability to remove excess fluid from the air spaces in these sheep was probably related more to a marked increase in paracellular permeability from injury, but it is possible that this insult led to significant damage to the alveolar epithelium and, subsequently, to a loss of salt and water transport capacity of alveolar epithelial cells.73
Interestingly, the expression and/or activity of ENaC and Na+-K+-ATPase were upregulated in multiple models of lung injury. In a subacute model of hyperoxia (85% O2 for seven days) Sznajder et al 74 were able to show an increase in active Na+ transport and lung liquid clearance at the end of the exposure period. Furthermore, this group of investigators has reported that enhanced Na+ transport is associated with augmented Na+-K+-ATPase activity in the alveolar type II cells of these animals at the end of hyperoxic exposure.75More recently Yue et al 76observed the increased expression and activity of sodium channels in alveolar type II cells of rats exposed to 85% O2 for seven days. Although these results suggest that Na+ transport mechanisms could be activated during hyperoxia induced lung injury, the proliferative response of alveolar type II cells seen in this model77 could also possibly explain such findings. Using a different model of lung injury (thiourea induced lung oedema) where there is no significant cell proliferation,78 we have also shown that lung Na+-K+-ATPase activity is increased during recovery from lung injury.79 The activity of the enzyme reached its maximum at 12 hours, a time when we start to observe a decrease in the oedema content of the lung.79This increased activity is also associated with an increased quantity of the enzyme in the lung and, more precisely, in alveolar type II cells at 12 hours.79 These results suggest that Na+ transport could be upregulated in lung injury.
However, other experimental data have suggested that the Na+ transport mechanism could be downregulated in severe lung injury. Firstly, it has recently been shown that inflammatory products such as reactive oxygen and nitrogen species can inhibit Na+ transport80 and Na+ channel activity.81 There are also conflicting data regarding the modulation of the expression and activity of Na+-K+-ATPase in an acute model of hyperoxic lung injury where animals are exposed to 100% O2 for 60 hours. Nici et al 82demonstrated increased expression of Na+-K+-ATPase mRNA and protein in exposed animals compared with controls. The same group has shown that this adaptive response of Na+-K+-ATPase in alveolar type II cells is associated with enhanced active Na+transport in vivo.83 However, Oliveraet al 84 recently reported a decrease in active Na+ transport at the end of hyperoxic exposure but with an increase at seven days after exposure. The difference between these findings can possibly be explained by a heterogeneous response to injury. In fact, recent data show that the response of the alveolar epithelium to this type of damage is quite heterogeneous and that the impact on the Na+ transport mechanism also depends on the severity of the injury.85
Collectively, these observations suggest that there could be significant activation of the Na+ transport mechanism in lung injury but this mechanism might not be functional when the alveolar epithelium is severely damaged.
HOW DO ALVEOLAR EPITHELIAL CELLS REPAIR INJURED AIR SPACES?
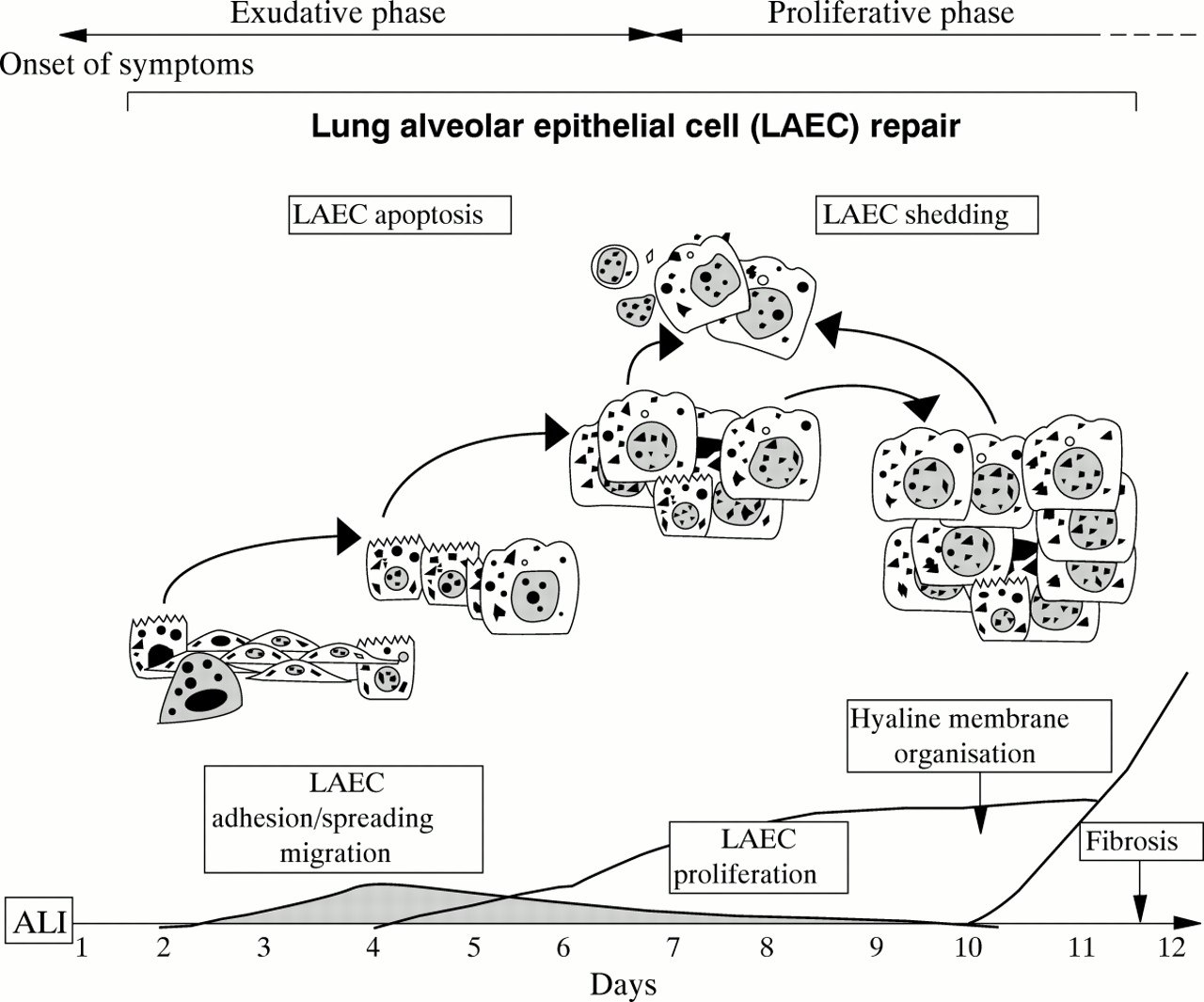
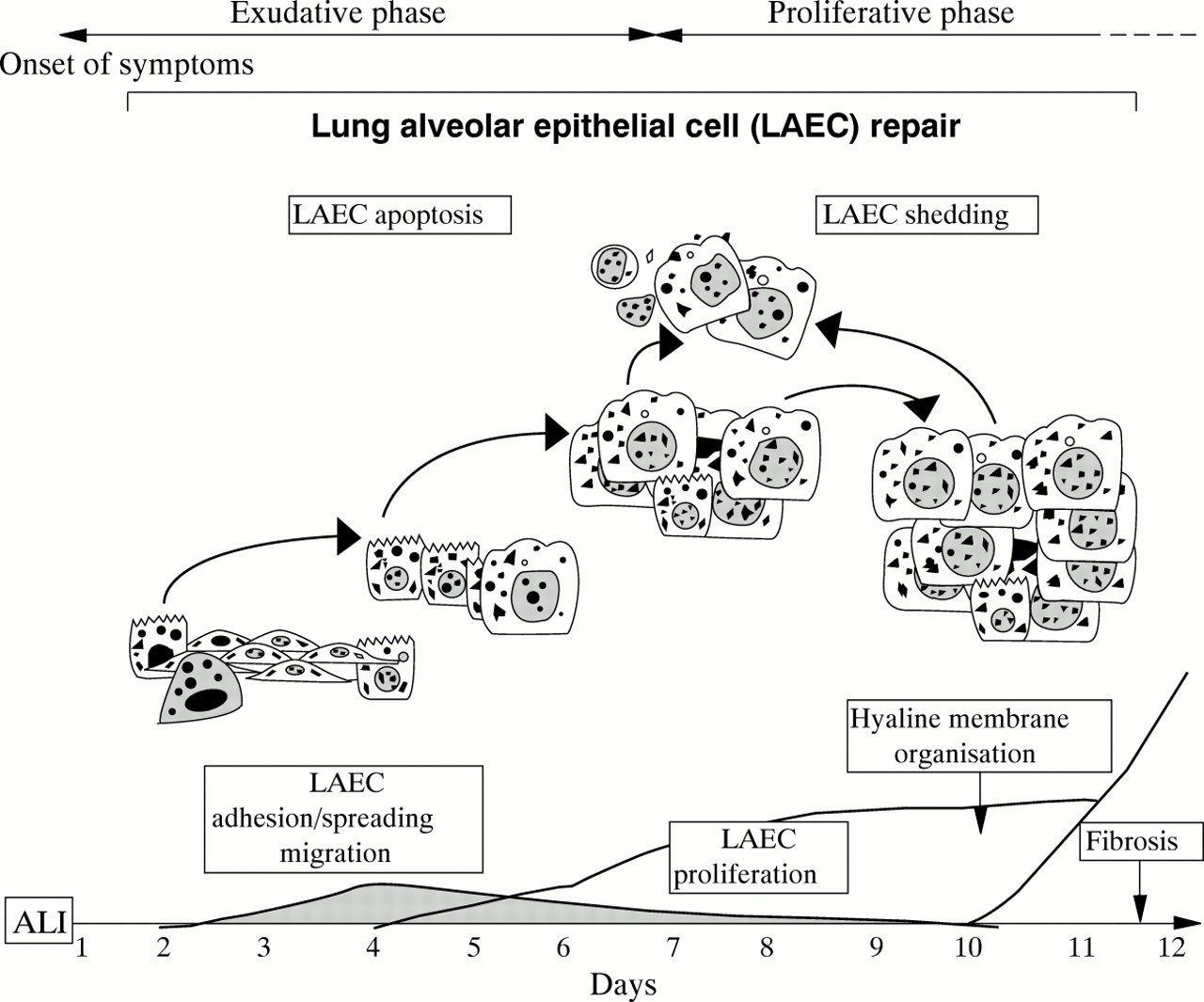
Although removal of oedema liquid is important in the resolution of ARDS, one of the most essential functions of the alveolar epithelium is participation in the repair of alveolar structures. Residual type II alveolar epithelial cells, which are more resistant to injury than type I alveolar epithelial cells, are the source of recovery for distal epithelia of the air spaces.17 The turnover rate of type II alveolar epithelial cells in the normal adult lung is remarkably low at about 4% per day but it is boosted after acute lung injury (ALI).14 ,15 ,17 ,86 ,87 However, proliferation of alveolar epithelial cells, which is the most obvious and easily measurable event leading to repair, needs several hours to take place and at least one or two days to be significant (fig2).14 ,15 ,86 Clearly, adhesion, spreading and migration are prerequisites to optimal alveolar epithelial cell repair (fig2).14 ,17 ,88 ,89 To date there is very little evidence of spreading-migration of alveolar epithelial cells in the lung in vivo. This can be explained by the lack of sensitive and specific tools for studying epithelial cell migration in vivo and also by the difficulty in realising real time observation of lung tissues in comparison with other tissues such as the skin, eyes and bowel.

Normal epithelial cell repair following acute lung injury. The different stages involved in the process are illustrated. ALI = acute lung injury; LAEC = lung alveolar epithelial cell.
There are at least two good reasons to consider migration-spreading-adhesion processes as being important for epithelial repair: (1) the increasing demonstration of early, efficient and sometimes sufficient locomotion and/or spreading of epithelial cells after tissue injury88-91 and (2) recent in vitro and ex vivo reports of induced alveolar epithelial cell migration-spreading under controlled conditions without proliferation.88-90 ,92 It is reasonable to postulate that, depending on the type and extent of injury (as it occurs in ARDS), migration-spreading, which is the fastest inducible repairing event, can proceed to restore or prepare and set up alveolar epithelial cell restitution by proliferation, as already observed in other organs to be resurfaced.93 ,94 Cell migration is a spatially and temporally integrated process with membrane extensions (for example, lamellipodia, filopodia), formation and stabilisation of cell substratum attachments, spreading of contractile forces and traction, coordination of rear release, and front edge progression.95 While this can take a couple of minutes for neutrophils or a couple of hours for fibroblasts, it needs a few hours to initiate but several hours to operate optimally (8–16 hours) for alveolar epithelial cells.88 Thus, it is likely that alveolar epithelial cell proliferation and migration overlap in some places, although there are arguments that both activities cannot coexist in the same cell.88 The two processes are complementary although each can be individually sufficient for wounding under specific conditions, with proliferation definitely being a powerful repair mechanism.15 ,17 ,86
Adhesion, spreading, migration and proliferation also require communication of the cells with their environment. Epithelial cells will interact with either a provisional matrix rich in fibronectin, fibrinogen and plasma proteins containing multifold or fragmented remnants of basal membranes,10 ,11 or with the extracellular matrix synthesised by the alveolar epithelial cells.96 Cell-matrix interactions are mediated by epithelial integrins through specific kinases such as pp125 FAK (focal adhesion kinase) and I-LK (integrin-linked kinase).97 Normal wound healing especially induces the expression of α5β1 (fibronectin receptor), and α3β1 (laminin receptor) as well as αν heterodimerisation with β3, β5 and β6 (vitronectin receptors) to a lesser degree.90 ,97 ,98 Interestingly, hyaline charged alveolar epithelial cells in areas of early diffuse alveolar damage with intra-alveolar fibrosis fail to express basal α5β1 and exhibit dissociated actin-like microfilaments with a pattern of partial detachment from the alveolar walls.99
In this respect epithelial cell shedding has been commonly observed for a long time in acute lung injury and chronic fibrosing alveolitis with a desquamative interstitial pneumonitis (DIP).100 ,101 Most of these processes could be retrospectively related to apoptosis (or programmed cell death) since loss of focal cell adhesion sites followed by matrix detachment can induce apoptosis.102 Furthermore, pathological investigations of acute diffuse alveolar damage have revealed upregulation of the apoptosis facilitators p53, WAF-1 (wild-type p53-activated fragment), and Bax in alveolar epithelial cells.103 ,104 Finally, Fas-Fas ligand interaction, one of the major systems controlling apoptosis of epithelial cells,105 can elicit widespread alveolar epithelial cell apoptosis with subsequent pulmonary fibrosis.106
Thus, it is reasonable to postulate that failure of alveolar epithelial cell adhesion-spreading-migration and excessive apoptosis can delay repair and favour fibrosis through the loss of natural inhibitory control exerted by epithelial-mesenchymal cross talk (fig3).14 ,15
Failure in epithelial cell repair following acute lung injury. The failure of migration and proliferation of alveolar epithelial cells leads to shedding of epithelial cells and the development of fibrosis. ALI = acute lung injury; LAEC = lung alveolar epithelial cell.
WHICH CYTOKINES ARE REQUIRED FOR ALVEOLAR EPITHELIAL CELL REPAIR?
Both migration and proliferation need modulators (table 1). Nearly all cytokines identified recently as promoters of alveolar epithelial cell migration are heparin sulphate binding proteins—for example, epithelial growth factor (EGF), transforming growth factor (TGF)α, keratinocyte growth factor (KGF), hepatocyte growth factor (HGF), fibroblast growth factor (FGF).86 KGF, HGF, and FGF are paracrine fibroblast derived polypeptides, while EGF and TGFα can act in an autocrine-paracrine manner on alveolar epithelial cells bearing receptors.86 ,107-112 These heparin binding cytokines are now well recognised and are clearly omnipresent in the lung during development107 ,108 and after lung injury,113-116 two processes that are associated with significant remodelling of the alveolar epithelium. KGF is a powerful promoter of proliferation and migration of alveolar epithelial cells110 ,117 and, when delivered locally, can reduce disease intensity and overall mortality in experimental models such as lung injury induced by acid, bleomycin, and hyperoxia.118-120 HGF can dramatically reverse Fas ligation induced fulminant hepatocyte apoptosis,121 which suggests that it could perform a similar function in the lung and then prevent fibroblast overgrowth. Finally, TGFα can stimulate alveolar epithelial cell wound closure in an in vitro model of alveolar type II cells.89
Cytokines affecting growth and motility of lung alveolar epithelial cells
The role of TGFβs in epithelial repair is complex and probably depends on the epithelium studied. Early after the induction of bleomycin lung injury there is a significant decrease in the secretion of biologically active TGFβ3.122 ,123Furthermore, following hyperoxia induced lung injury, not only is biologically active TGFβ reduced,123 but the expression of TGFβ receptors I and II is also diminished.124 Since TGFβ is a powerful growth inhibitor of cells of epithelial origin, this initial nadir of TGFβ3 after lung injury could potentially help and allow alveolar epithelial cell proliferation and repair. TGFβs also upregulate the production of matrix protein components125 and are important regulators of integrin expression on epithelial cells,126 ,127 two physiological phenomena that are vital in the repair process, particularly for cell migration.
Platelet-derived growth factor (PDGF) is an intriguing putative modulator for alveolar epithelial cell repair. PDGF-BB is present early in increased amounts in injured lungs128 and has long been designated a culprit in lung fibrogenesis.129 However, PDGF is also mitogenic for fetal alveolar epithelial cells.130 Since it can improve wound healing in the skin131 and gastric epithelial cell restoration,132 it would be interesting to study migration as well as proliferation of alveolar epithelial cells in the injured lung in response to PDGF.
Lymphomonokines IL-2, IL-15 and IFN-γ may play an unexpected role in epithelial restitution in ARDS. In this respect, IL-2 and the functionally related IL-15 have already been clearly identified as major factors involved in intestinal epithelial cell repair.94 ,133 ,134 IFN-γ is also known to decrease keratinocyte migration and alveolar epithelial cell growth and locomotion.88 ,135 ,136 On the other hand, IFN-γ increases IL-2R expression on alveolar epithelial cells135and promotes IL-2 induced alveolar epithelial cell growth activity.88 ,134 While IL-2 and IL-15 are already recognised protectors against lymphocyte and neutrophil apoptosis,137-139 IL-15 can also dramatically reverse fulminant hepatocyte apoptosis140 and could exert similar anti-apoptotic protection in alveolar epithelial cells.
Rescue therapy of the alveolar epithelium
Since the alveolar epithelium is so important for lung function, and based on the knowledge acquired over the past few years, we can imagine therapeutic approaches aimed at the alveolar epithelium that could accelerate recovery from ARDS.
STRATEGIES TO ACCELERATE OEDEMA CLEARANCE
Even if active Na+ transport is involved in the resolution of pulmonary oedema, could we stimulate it to enhance oedema resolution? In one clinical study of patients with lung injury it was shown that about 40% of subjects were able to reabsorb some of the alveolar oedema fluid within 12 hours of intubation.9These patients recovered more rapidly from respiratory failure and also had lower mortality. In contrast, patients who did not reabsorb any alveolar oedema fluid in the first 12 hours after acute lung injury had protracted respiratory failure and higher mortality.9Based on clinical studies, the ability of the alveolar epithelial barrier to reabsorb alveolar oedema fluid within the first 12 hours after acute lung injury is preserved in 30–40% of patients.9 The results suggest that pharmacological manipulation of the alveolar epithelium could possibly accelerate the resolution of pulmonary oedema in zones where it is intact. In other areas of the lung, where significant injury to the alveolar epithelium has occurred, reconstitution will be necessary before this process can be modulated. Finally, in areas of intense alveolar type II cell proliferation it is possible that there would be intrinsic enhanced alveolar liquid clearance (fig 4).141

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic diagram illustrating three potential alveolar environments that can be encountered in an injured lung. In some alveoli there is significant damage to the alveolar epithelium. In these regions alveolar epithelial repair will be needed before stimulation of ion transport can be achieved. Other alveoli will maintain normal alveolar function. In these alveoli pharmacological stimulation of alveolar liquid clearance will be possible. Finally, in other areas of the lung there will be a proliferation of alveolar type II cells which will lead to an intrinsic enhanced clearance rate of oedema liquid.
What are the pharmacological agents that could be used to stimulate clearance of lung liquid in patients with an intact alveolar epithelium? The agents most studied at the moment in animal models have been the β adrenergic agonists which can stimulate clearance of lung liquid in several species20 ,22 ,23 including the ex vivo human lung.125 ,142 This effect of the β adrenergic agonists can be obtained either by intra-alveolar or intravenous administration.20 More recently it was also shown that aerosolised salmeterol, a lipid soluble β2 agonist, can stimulate clearance of lung liquid at a clinically relevant dose.143
Not only can the β adrenergic agonist stimulate clearance of lung liquid in the normal lung, but it can also accelerate liquid clearance in the presence of hyperoxic lung injury. Lasnieret al 144 reported that terbutaline (10–3 M) increased alveolar liquid clearance by 50% in rats exposed to 100% oxygen for 60 hours. Similarly, Garatet al 145 noted that alveolar liquid clearance was enhanced by 45% in animals treated with terbutaline (10–4 M) after 40 hours exposure to 100% oxygen. Furthermore, the β adrenergic agonist not only modulates the Na+ channel or Na+-K++ATPase activity involved in this response, but it can also alter the expression or quantity of these proteins (fig 2). In fact, some recent studies have shown that the sustained treatment of alveolar type II cells with β adrenergic agonists can increase the expression of ENaC and Na+-K+-ATPase.52 Thus, it is possible that sustained treatment with β adrenergic agonists could play a part in the management of patients with lung injury. Furthermore, other vasoactive agents commonly used in patients in the intensive care unit could have therapeutic applications—for example, dobutamine146 and dopamine147 have recently been shown to upregulate alveolar liquid clearance.
However, controlled clinical trials will be needed to evaluate these potential treatments since clinical conditions in which they would be used could vary significantly from one patient to another, and this could have significant repercussions on their efficacy. As we have already discussed, mild septic shock is associated with an increase in lung liquid clearance secondary to the endogenous release of catecholamines.72 However, more severe sepsis is accompanied by significant dysfunction of the alveolar epithelium, preventing proper modulation of lung liquid clearance.73Furthermore, recent work has indicated that, after prolonged haemorrhagic shock in rats, oxidant mechanisms downregulate the response of the alveolar epithelium to β2 agonist stimulation.148 Hence, it is unlikely that in severe lung injury the use of β adrenergic agonists or vasoactive agents will be effective as a single therapy for rescue of the alveolar epithelium. Finally, as has already been shown in septic rats, there is an endogenous response to this stress by the release of endogenous catecholamines,72 ,148 but it is still uncertain if exogenous β adrenergic agents given under such circumstances would add to the endogenous release. Clearly, the idea of therapeutically modulating the alveolar epithelium to enhance liquid clearance is feasible, but many obstacles remain to be overcome before the concept can be tested at the bedside.
STRATEGIES TO ACCELERATE EPITHELIAL REPAIR
Although multiple cytokines have been shown to be important in repair of the alveolar epithelium, few studies have been done to evaluate their potential in the treatment of lung injury. Recent animal experiments indicate that HGF and KGF could have a significant role as new therapeutic agents. Administration of HGF to animals with lung injury can stimulate alveolar epithelial cell proliferation.149 Furthermore, pretreatment of rats with KGF (5 mg/kg) prior to lung injury induced by thiourea, bleomycin or hyperoxia leads to a decrease in the severity of lung injury.118-120 However, it is still unclear if this protective effect of KGF is linked to its impact on the proliferation of alveolar epithelial cells. In fact, KGF is not only known to modulate the phenotypic characteristic of alveolar epithelial cells117 but is also able to increase Na+-K+-ATPase expression and upregulate Na+ transport in cultured alveolar type II cells.150 Furthermore, in vivo experiments performed recently to explore the mechanism responsible for this response suggest that the protective effect of KGF includes not only the heightened proliferation of alveolar type II cells but also the upregulation of Na+ transport across the alveolar epithelium.151 The potential value of manipulating therapeutically other growth factors such as TGFα or TGFβ in lung injury has not yet been evaluated.
Although growth factors could have some therapeutic potential in ARDS, much work is needed to define better their therapeutic value since manipulation of this system could also have a negative impact. In fact, it is known that overexpression of certain growth factors could lead to pulmonary fibrosis. Indeed, transgenic mice overexpressing TGFα develop pulmonary fibrosis.152 Thus, as with the development of therapeutic strategies for enhancing oedema clearance, strategies to promote epithelial repair are still in their embryonic stage of development.
Conclusion
Treatment of ARDS at the moment remains mainly supportive since none of the therapies evaluated has been shown to reduce morbidity or mortality in controlled clinical trials. Nevertheless, in recent years standard supportive therapies seem to have led to a significant decrease in mortality. A further fall in mortality from ARDS should evolve from a better understanding of the causes of death in patients with ARDS.
Since the major causes of death in ARDS have been shown to be non-pulmonary organ dysfunction,1 it is relevant to wonder why a treatment aimed at the alveolar epithelium would help these patients. Although the major source of mortality is non-pulmonary organ dysfunction, it is possible that lung dysfunction could be significantly implicated in this process. In fact, pulmonary dysfunction leads to significant abnormalities in gas exchange which necessitate mechanical ventilation. There is an increasing amount of evidence that mechanical ventilation of the injured lung153 not only causes further injury but also induces a systemic inflammatory response that could be important for the development of non-pulmonary organ dysfunction.154Acceleration of oedema clearance and repair of the alveolar epithelium should enhance the resolution of respiratory failure and decrease the time patients have to be on mechanical ventilation. A combination of treatments aimed at the control of the inflammatory response and at the restitution of normal epithelial function should lead eventually to a fall in the mortality of patients with ARDS.
Acknowledgments
This work was supported in part by The Medical Research Council of Canada (grant MT-1203). Dr Berthiaume and Dr Lesur are Chercheur-Boursier clinicians from Fonds de la Recherche en Santé du Québec.