Article Text

Statistics from Altmetric.com
Neutrophil influx into the extravascular compartments of the lungs is a defining characteristic of the Acute Respiratory Distress Syndrome (ARDS).1 During ARDS, circulating neutrophils become primed, resulting in reduced deformability and retention within the pulmonary capillary bed2 ,3 followed by migration across the endothelium, through the interstitium and across the epithelium into the airspaces.4 As neutrophils migrate, they may become activated to phagocytose invading pathogens and release oxidants, proteases and neutrophil extracellular traps, all of which play a role in killing pathogens. Although neutrophils may migrate into the airspaces without inducing an increase in protein permeability under certain conditions,5 in ARDS neutrophils and their toxic mediators can cause tissue injury, including an increase in lung epithelial and endothelial permeability6–9 which leads to the influx of protein-rich alveolar oedema and arterial hypoxaemia.10 In fact, mortality from ARDS correlates with the extent of neutrophilia in the lung.1 Treatment for ARDS is mainly supportive, consisting of low tidal volume ventilation and fluid restriction, approaches which have substantially improved outcomes. However, to further improve outcomes, specific therapies to limit inflammatory lung injury while preserving host defense are needed. Therefore, it is critical that we understand the mechanisms regulating neutrophil recruitment, priming, activation and effector functions.
Leucocyte chemokines are classified into several families based on the position of cysteine (C) residues: CXC, CC, C and CX3C.11 Neutrophil migration has classically been thought to be driven by the CXC chemokines, with CXCL8 (interleukin 8) in humans and its orthologs CXCL1 (KC) and CXCL2 (MIP2) in mice being the prototypical neutrophil chemokines.12 CXC chemokines are elevated in patients with ARDS13 and animal models of lung injury,7 ,12 and CXCL8 levels are predictive of disease development,14 severity15 and mortality.16 However, although CXC chemokines are responsible for much of the neutrophil recruitment to the lungs during lung injury, blockade of CXCL8 or CXCL1/2 prevents some but not all neutrophil recruitment.7 ,12 Additional CXC chemokines such as CXCL5 (LIX/ENA-78), CXCL12 (SDF-1) and CXCL15 (lungkine), as well as other mediators such as LTB4 and C5a, also are neutrophil chemoattractants.8
Chambers et al have previously published elegant animal studies demonstrating that the CC chemokines CCL2 (MCP-1) and CCL7 (MCP-3), classically chemotactic for monocytes,11 contribute to neutrophil recruitment in acute lung injury. After challenge with lipopolysaccharide (LPS) or live bacteria, CCL2 and CCL7 were rapidly upregulated in the lungs, the CC chemokine receptors CCR1 and CCR2 were expressed on neutrophils, and blockade of CCL2 and/or CCL7 attenuated neutrophil recruitment.17 ,18
In Thorax, Williams et al19 tested their experimental findings in patients, asking whether CCL2 and CCL7 are similarly important in neutrophil recruitment in ARDS. Both CCL2 and CCL7 were elevated in the bronchoalveolar lavage (BAL) fluid of patients with ARDS and the neutrophil chemotactic activity of BAL fluid was as much attributable to either CCL2 or CCL7 as to CXCL8. Although CCL2 and CCL7 were weaker neutrophil chemoattractants than CXCL8 when chemotaxis towards recombinant chemokines was tested, CCL2 and CCL7 each significantly enhanced neutrophil chemotaxis to CXCL8, suggesting that the effect is synergistic. Finally, their studies demonstrated that the CXC chemokine receptor CXCR1 was downregulated while the CC chemokine receptor CCR2 was upregulated as neutrophils migrated from the bloodstream into the airspaces (figure 1).

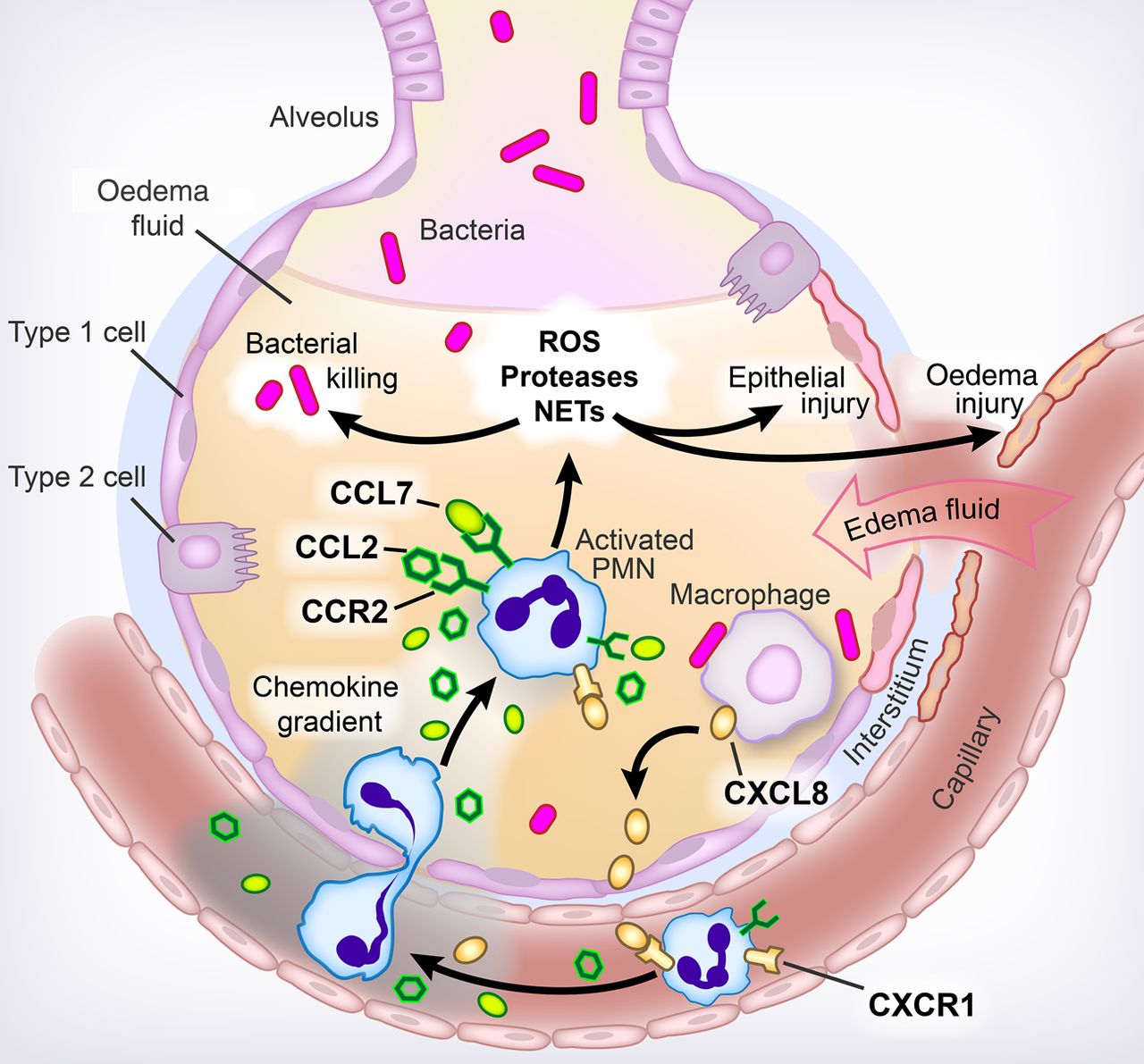{kind=link}
Neutrophil influx in ARDS. In patients with acute lung injury from ARDS, resident lung cells such as macrophages secrete chemokines that induce neutrophils or polymorphonuclear cells (PMN) to migrate from the bloodstream into the airspaces. Classically, neutrophil migration has been thought to be driven by CXC chemokines, with CXCL8 being the protypical neutrophil chemokine, inducing migration by acting on its receptors, mainly CXCR1 in humans. Chambers et al have demonstrated that two members of the CC family of chemokines, CCL2 and CCL7, are upregulated in the lungs of patients with ARDS. Their receptor, CCR2, is upregulated on neutrophils as they migrate from the vasculature into the airspaces, while CXCR1 is downregulated. Moreover, these CC chemokines act in synergy with CXCL8 to induce neutrophil influx. As neutrophils migrate into the lungs, they become activated, releasing toxic mediators such as reactive oxygen species (ROS), proteases and neutrophil extracellular traps (NETs), which function to kill invading pathogens but also may cause epithelial and endothelial injury, leading to the development of protein-rich alveolar oedema. Figure prepared by Diana Lim.
This study was rigorously performed, leading to well justified conclusions. The findings are consistent with prior literature demonstrating that CCL2 and CCL7 can function as neutrophil chemoattractants,20 ,21 including in models of lung injury,22 ,23 and are elevated in human ARDS.13 ,24 However, the new evidence that the neutrophil chemotactic activity in BAL fluid from patients with ARDS is highly attributable to CCL2 and CCL7 and that CCL2/7 potentiates the chemotactic activity of CXCL8 is novel and important. Moreover, given that the neutrophils used in the chemotaxis assays were isolated from the blood of healthy volunteers, which likely express less CCR2 than those actively migrating into the lungs during ARDS, the role of CCL2 and CCL7 in neutrophil chemotaxis in ARDS may even be underestimated.
These studies raise several mechanistic questions. First, the extent to which CCL2/7 are directly chemotactic for neutrophils is unclear, as the degree of neutrophil chemotaxis induced by recombinant CCL2 or CCL7 was small and did not reach statistical significance, even with concentrations of recombinant chemokines much higher than those detected in the BAL fluid of patients with ARDS. Still, reasonable concentrations of CCL2/7 do enhance the chemotactic potency of CXCL8. While the authors speculate that enhanced receptor occupancy may contribute to this synergistic effect, future studies should explore the mechanism underlying this effect as well as whether CCL2/7 also acts in synergy with other CXC (or CC) chemokines. In addition, the cellular sources of CCL2 and CCL7 and the mechanisms that induce their synthesis are not completely understood. Reports suggest the alveolar and bronchial epithelium as likely sources,17 ,23 ,25 and expression appears to be regulated by proteinase-activated receptors.17 ,18 In addition, while the authors have demonstrated that CCL2/7 are directly chemotactic for neutrophils, there may be additional mechanisms through which CCL2/7 induce neutrophil recruitment, such as via activation of resident alveolar macrophages or epithelial cells to secrete CXC chemokines or by inducing monocyte recruitment, which in turn may facilitate neutrophil recruitment.26 In addition, investigation of the extent to which these CC chemokines induce chemotaxis and prime and activate neutrophils and prolong neutrophil life span, as CXCL8 can, is warranted. Along these lines, additional studies should be done to determine whether blockade of these chemokines also mitigates lung injury (permeability) in human ARDS models, as is the case with CXCL8 blockade.7 Finally, the effect of CCL2/7 inhibition, with associated blockade of neutrophil influx, on the ability of the lungs to clear bacteria should be tested in ARDS models of live bacterial infection.
The holy grail of ARDS therapy would be to limit neutrophil recruitment and priming/activation while preserving host defence. However, neutrophil function is complex, requiring migration, priming and activation, each potentially induced by a variety of mediators, for their full microbicidal and cytotoxic effects. A better understanding of the precise role of each mediator is a prerequisite for the design of novel therapies that would intervene at one or more steps to limit tissue damage while preserving microbicidal function. In this context, this manuscript has made an important contribution.
Because ARDS is heterogeneous, it is likely that therapies will need to be personalised to the individual patient. In the patients with ARDS studied here, there was substantial variability in chemokine levels, the extent to which each chemokine accounted for the overall chemotactic activity of the BAL fluid, and neutrophil expression of CXC and CC chemokine receptors. Host factors surely contribute, but the specific aetiology of ARDS is likely to be a critical determinant of the specific chemokine-receptor pathways involved. In mice, both CCL2 and CCL7 were important neutrophil chemokines in the LPS model, while in the Streptococcus pneumoniae model, CCL7 but not CCL2 was implicated.17 Whether different neutrophil chemokines are important in human ARDS of different aetiologies remains to be determined. It would also be interesting to know whether the levels and relative chemotactic activity of the CC chemokines, or expression of their receptors, correlates with neutrophil numbers, disease severity or prognosis, as is the case for CXC chemokines.15 ,16 Finally, neutrophils are increasingly recognised to be heterogeneous in terms of cell surface marker expression and function;27 different subsets of neutrophils may be differentially responsive to CC and CXC chemokines.
A targeted and personalised approach to inhibit one or more mediators of neutrophil function while permitting the activity of others could theoretically mitigate neutrophil-dependent tissue damage without impairing host defense. However, because the mediators that induce neutrophil chemotaxis and activation are complex, redundant and pathogen-dependent, targeting neutrophils is still likely to be challenging as a basic treatment for ARDS, especially once the tissue injury is established. Indeed, clinical trials of agents directed towards reducing neutrophil migration and/or activation have not improved outcomes.28 Ultimately, a multifaceted approach aimed at attenuating inflammatory tissue injury while preserving host defense as well as promoting the resolution of inflammation,29 repair of the injured lung30 and the reabsorption of oedema fluid is likely to be necessary.
Acknowledgments
The authors thank Diana Lim for her work in preparing figure 1.
References
Footnotes
Funding RLZ: NIH HL131608, Boettcher Foundation, American Heart Association, University of Colorado Department of Medicine, MAM: NHLBI HL51856, NHLBI HL51856.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.
Linked Articles
- Respiratory research
- Airwaves