Article Text

Statistics from Altmetric.com
Historical introduction
Salicylic acid, the active substance in plants used for thousands of years as medicaments, was synthesised by Kolbe in Germany in 1874. MacLagan1 and Stricker2showed that it was effective in rheumatic fever. A few years later sodium salicylate was also in use as a treatment for chronic rheumatoid arthritis and gout as well as an antiseptic compound.
Felix Hoffman was a young chemist working at Bayer. Legend has it that his father, who was taking salicylic acid to treat his arthritis, complained to his son about its bitter taste. Felix responded by adding an acetyl group to salicylic acid to make acetylsalicylic acid. Heinrich Dreser, the Company's head of pharmacology, showed it to be analgesic, antipyretic, and anti-inflammatory.3 Bayer introduced the new drug as “aspirin” in 1899 and sales have increased ever since.
In the latter part of the 20th century several other non-steroidal anti-inflammatory drugs (NSAIDs) were discovered, including antipyrine, phenacetin, phenylbutazone and, more recently, the fenamates, indomethacin and naproxen. Despite the diversity of their chemical structures, these drugs all share the same therapeutic properties. They alleviate the swelling, redness and pain of inflammation, reduce a general fever, and cure a headache. They also share, but not equally, a number of side effects including damage to the gastric mucosa, delay in the birth process, and damage to the kidney. A particularly interesting “side effect”, now used prophylactically, is the anti-thrombotic effect. Many clinical trials have shown that aspirin given once a day in doses as low as 75 mg will help to prevent heart attacks or strokes.
When a chemically diverse group of drugs shares not only the same therapeutic qualities (which in themselves have not much connection with each other), but also the same side effects, it is a fairly safe bet that their actions are based on a single biochemical intervention. For many years pharmacologists and biochemists searched for such a common mode of action without finding one that was satisfactory.
It was against this background that, using a crude preparation of prostaglandin synthase (now known as cyclo-oxygenase or COX), Vane4 found a dose dependent inhibition of prostaglandin formation by aspirin, salicylate, and indomethacin but not by morphine. Two other reports from the same laboratory lent support to his findings. Smith and Willis5 showed that aspirin prevented the release of prostaglandins from aggregating human platelets and Ferreira et al 6 demonstrated that aspirin-like drugs blocked prostaglandin release from the perfused, isolated spleen of the dog. Vane4 proposed that all NSAIDs act by inhibiting COX, thereby reducing prostaglandin formation, providing a unifying explanation for their therapeutic actions and their side effects. This also firmly established certain prostaglandins as important mediators of inflammatory disease (see reviews by Vane and Botting7 and Vaneet al 8). COX first cyclises arachidonic acid to form prostaglandin (PG) G2 and the peroxidase part of the enzyme then reduces PGG2 to PGH2.
Discovery of COX-2
Over the next 20 years several groups postulated the existence of isoforms of COX. Then Rosen et al,9 studying COX in epithelial cells from the trachea, found an increase in activity of COX during prolonged cell culture. This increase in activity was not accounted for by an increase in the known mRNA of 2.8 kb. They found a second mRNA of 4.0 kb and suggested that this was derived from a distinct COX-related gene which encoded for a protein with COX activity. Needleman and his group10-12 reported that bacterial lipopolysaccharide (LPS) increased the synthesis of prostaglandins in human monocytes in vitro and in mouse peritoneal macrophages in vivo. This increase, but not the basal level of enzyme, was inhibited by dexamethasone and associated with de novo synthesis of new COX protein. This gave rise to the concept of “constitutive” and “inducible” forms of COX.
The breakthrough came from molecular biologists outside the field of prostaglandins. Simmons et al,13 studying neoplastic transformation, discovered a second form of COX induced by v-src, serum, or phorbol esters in chicken embryo cells.13 ,14 It was encoded by a 4.1 kb mRNA similar in size to that reported by Rosen et al.9 They cloned the gene, deduced the protein structure, and found it homologous to COX but to no other known protein. Herschman and colleagues15 independently found a similar gene in the mouse, as did Simmons et al,16 O'Banion et al,17 and Sirois and Richards.18
COX-1 and COX-2 have molecular weights of 71 kd and a 60% homology. Glucocorticoids inhibit the expression of COX-2 and this is an additional aspect of their anti-inflammatory action. The levels of COX-2, normally very low in cells, are tightly controlled by a number of factors including cytokines, intracellular messengers, and by the availability of substrate.
COX-1
COX-1 has three folding units: an epidermal growth factor-like domain, a membrane binding section, and an enzymatic domain. The sites for peroxidase and cyclo-oxygenase activity are adjacent but spatially distinct. The enzyme integrates into only a single leaflet of the membrane lipid bilayer and thus the position of the COX channel allows arachidonic acid to gain access to the active site from the interior of the bilayer.19
NSAIDs compete with arachidonic acid for binding to the active site, thereby excluding access for the substrate.20 Uniquely, aspirin irreversibly inhibits COX-1 by acetylation of serine 530. COX-1 has clear physiological functions. Its activation leads, for instance, to the production of prostacyclin which, when released by the endothelium is anti-thrombogenic,21 and when released by the gastric mucosa is cytoprotective.22
COX-2
The structure of COX-223 closely resembles that of COX-1 but, fortunately for the medicinal chemist, the binding site for arachidonic acid (the COX channel) is slightly different. The active site of COX-2 is a little larger and can accommodate bigger structures than that of COX-1. A secondary internal pocket contributes significantly to the larger volume of the active site of COX-2, although the central channel is also 17% bigger.
Since COX-2 is induced by inflammatory stimuli and by cytokines in migratory and other cells, it is attractive to suggest that the anti-inflammatory actions of NSAIDs result from the inhibition of COX-2, whereas the unwanted side effects such as damage to the stomach lining are caused by inhibition of the constitutive enzyme, COX-1. This general concept is now widely accepted.
Functions of COX-1 and COX-2
LUNGS
Prostacyclin is a potent vasodilator of the pulmonary circulation in humans and other species. This endothelium derived prostacyclin is well placed to function as a local vasodilator and to prevent the formation of microthrombi.24
Pulmonary blood vessels are constricted by PGF2α and thromboxane (TX) A2 but, in some species, they are dilated by PGE2. The vasoconstrictor responses to PGF2α are potentiated by hypoxia. Mediators of inflammation such as bradykinin, histamine, and 5-hydroxytryptamine all release prostaglandins from lung tissue. Histamine releases PGF2α from human lung fragments by stimulating H1 receptors. The lungs of asthmatic subjects produce more histamine than normal lungs, which correlates with the greater number of mast cells found in asthmatic lungs.25
The airways of most species, including humans, are constricted by PGF2α, TXA2, PGD2, and PGI2 whereas PGE2 is weakly bronchodilator. Asthmatic subjects are 8000 times more sensitive to the bronchoconstrictor action of inhaled PGF2α than healthy subjects. COX-2 expression in airways has been reviewed by Barneset al.26 Airway hyperreactivity, a feature of allergic asthma, is associated with inflammation of the airways. There is increased expression of COX-2 mRNA and of enzyme protein with no change in COX-1 levels in pulmonary epithelial cells, airway smooth muscle cells, pulmonary endothelial cells, and alveolar macrophages treated with LPS or proinflammatory cytokines. In the carrageenin induced pleurisy model of inflammation, levels of COX-2 protein increased maximally at two hours in the cell pellets of pleural exudate.27 However, lung tissue can also express COX-2 constitutively. COX-2 mRNA is weakly expressed in unstimulated rat isolated perfused lungs and is upregulated by nitric oxide (NO) donors.28 Human lungs obtained from accident victims29 and human cultured pulmonary epithelial cells expressed more constitutive COX-2 than constitutive COX-1.30 ,31 Interestingly, hypoxia induces COX-2 gene expression in isolated perfused lungs of the rat without affecting the mRNA for COX-1.28 This induction of the COX-2 gene by hypoxia was inhibited by NO donors, which may represent one of the mechanisms of the beneficial effect of inhaled NO in pulmonary hypertension.
Inflammatory stimuli cause differential release of prostaglandins from various regions of the lungs. Human cultured pulmonary epithelial cells stimulated with LPS, interleukin (IL)-1α, tumour necrosis factor (TNF)α, or a mixture of cytokines synthesise mainly PGE2together with smaller amounts of PGF2α, PGI2, and TXA2. This prostaglandin production can be suppressed by dexamethasone32 which indicates that it is caused by COX-2.
Pollutants such as those in car exhausts can also induce COX-2 in human cultured airway epithelial cells, resulting in an increased formation of PGE2 and PGF2α.33PGE2 and TXB2, in addition to COX-2 protein, could be detected in the inflammatory exudate produced by injection of carrageenin into the rat pleural cavity.34 Thus, it seems likely that COX-2 is upregulated in the inflamed lungs of asthmatic patients resulting in increased production of bronchoconstrictor prostaglandins which exert an exaggerated effect on the bronchiolar smooth muscle that has become hyperreactive to constrictor agents.
ENDOTHELIUM
Endothelial cells generate prostacyclin which is anti-aggregatory and a vasodilator. They clearly contain COX-1 but McAdamet al 35 and Catella-Lawsonet al,36 using the selective COX-2 inhibitors celecoxib or rofecoxib in volunteers, found that the urinary excretion of the prostacyclin metabolite was substantially suppressed by anti-inflammatory doses of either drug. Thus, in the endothelium prostacyclin is mainly produced by COX-2, probably induced by laminar shear stress.37
GASTROINTESTINAL TRACT
The “cytoprotective” action of prostaglandins in preventing gastric erosions and ulceration is mainly brought about by endogenously produced prostacyclin and PGE2 which reduce gastric acid secretion, exert a direct vasodilator action on the vessels of the gastric mucosa, and stimulate the secretion of viscous mucus and duodenal bicarbonate.38 In most species, including humans, the protective prostaglandins are synthesised by COX-1, although COX-2 has been reported in the normal rat stomach,39 in human gastric mucosa infected with Helicobacter pylori,40 and in ulcerative colitis.41COX-2 is also expressed around the periphery of gastric ulcers in mice and rats. It may be involved in wound healing.42-44 Large quantities of COX-2 are expressed in experimentally induced and in human colon cancers.45 ,46
KIDNEY
Maintenance of kidney function both in animal models of disease states and in patients with congestive heart failure, liver cirrhosis, or renal insufficiency is dependent on vasodilator prostaglandins. These patients are therefore at risk of renal ischaemia when prostaglandin synthesis is reduced by NSAIDs. Synthesis of PGE2 and prostacyclin is mainly by COX-1, although there are discrete cells in the macula densa that contain constitutive COX-2.47 ,48 Prostacyclin made by COX-2 may drive the renin-angiotensin system48; Schneider and Stahl49 have reviewed this rapidly evolving field.
Mice that lack the gene for production of COX-1 appear to be healthy and do not show significant signs of kidney disease. This is in accord with the finding that inhibition of COX-1 by NSAIDs does not materially alter renal function under normal physiological conditions, although a small percentage of those taking NSAIDs have mild fluid retention. However, Morham et al reported that the kidneys failed to develop fully after birth in COX-2(–/–)null mice, with the result that the animals died before they were eight weeks old.50 More recently, after some backcrossing of the same COX-2(–/–) deficient mice in a different laboratory, a breed of COX-2(–/–) mice has been achieved which has little or no kidney malfunction and which live for three years.51
Fitzgerald's group36 compared the renal effects of the non-selective COX inhibitor indomethacin with those of the COX-2 inhibitor rofecoxib and with placebo in healthy older adults over two weeks. Both active regimes were associated with a transient but significant decline in urinary sodium excretion during the first 72 hours. The glomerular filtration rate (GFR) was decreased by indomethacin but not changed significantly by rofecoxib. Thus, acute sodium retention by NSAIDs in healthy adults is mediated by inhibition of COX-2, whereas depression of GFR is caused by inhibition of COX-1.
CENTRAL NERVOUS SYSTEM
COX-1 is found in neurones throughout the brain but it is most abundant in the forebrain where prostaglandins may be involved in complex integrative functions such as control of the autonomic nervous system and in sensory processing. COX-2 mRNA is induced in brain tissue and in cultured glial cells by pyrogenic substances such as LPS, IL-1, or TNF.52 However, low levels of COX-2 protein and COX-2 mRNA have been detected in neurones of the forebrain without previous stimulation by proinflammatory stimuli. These “basal” levels of COX-2 are particularly high in neonates and are probably induced by physiological nervous activity. Intense nerve stimulation, leading to seizures, induces COX-2 mRNA in discrete neurones of the hippocampus,53 whereas acute stress raises levels in the cerebral cortex. COX-2 mRNA is also constitutively expressed in the spinal cord of normal rats and may be involved with processing of nociceptive stimuli.54
Endogenous fever producing PGE2 is thought to originate from COX-2 induced by LPS or IL-1 in endothelial cells lining the blood vessels of the hypothalamus.52 Li et al 55 tested the effects of LPS in producing a fever in knockout mice. Wild type mice and COX-1(+/–) and COX-1(–/–) mice all responded to LPS with a 1°C rise in core temperature within one hour: the fever gradually abated over the next four hours. By contrast, COX-2(+/–) and COX-2(–/–) mice displayed no temperature rise after LPS. Thus, COX-2 is necessary for LPS induced fever production. A corollary of this finding is that there is unlikely to be a COX-3 through which paracetamol brings down a fever. The selective COX-2 inhibitor rofecoxib is a potent antipyretic agent in man.56
REPRODUCTIVE SYSTEM
Expression of COX-1 is much greater than that of COX-2 in the fetal heart, kidneys, lungs, and brain, as well as in the decidual lining of the uterus.57 ,58 Prostaglandins synthesised by COX-1 are apparently essential for the survival of fetuses during parturition, since the majority of offspring born to homozygous COX-1 knockout mice do not survive.59 This high mortality may result from the premature closure of the ductus arteriosus. Female COX-2 knockout mice are mostly infertile, producing very few offspring because of a reduction in ovulation.60 COX-2 induction is involved in ovulation and is clearly the trigger for parturition,61 ,62 leading to PGF2α release to cause contractions of the uterine smooth muscle.
Selective inhibition of COX-2
An estimated 34–46% of patients on chronic NSAID treatment will have some form of gastrointestinal adverse event. In the USA some 100 000 patients on NSAIDs are admitted to hospital each year because of perforations, ulcers, or bleeding in the stomach (PUBs).63 Some 15% of these patients die in intensive care. Of course, these hospital admissions only represent the extreme of gastric irritation, which ranges from mild dyspepsia all the way through to PUBs. Even ibuprofen, recognised as one of the mildest gastric irritants, causes problems in a significant proportion of patients. Clearly, there is a dramatic need for anti-inflammatory drugs that do not affect the stomach.
COX-2/COX-1 RATIOS
The importance of the discovery of inducible COX-2 is highlighted by the differences in pharmacology of the two enzymes.64Aspirin, indomethacin, and ibuprofen are much less active against COX-2 than against COX-1.65 Indeed, the strongest inhibitors of COX-1 such as aspirin, indomethacin, and piroxicam are the NSAIDs that cause the most damage to the stomach.
There are now many methods for measuring COX-2/COX-1 ratios, varying from human enzyme assays in vitro66 to assays in human blood samples.67-69 In general, the whole blood assays are reproducible from laboratory to laboratory and provide conditions that approach the physiological. For instance, any plasma protein binding of the drug will automatically be taken into account.
Implicit in the theory that COX-2 causes inflammation is that NSAIDs are used in inflammation because they inhibit COX-2. The range of activities of NSAIDs against COX-1 compared with COX-2 explains the variations in the side effects of NSAIDs at their anti-inflammatory doses. Garcia Rodriguez and Jick70 have published a comparison of epidemiological data on the side effects of NSAIDs. Piroxicam and indomethacin in anti-inflammatory doses showed high gastrointestinal toxicity. These drugs have a much higher potency against COX-1 than against COX-2.7 Thus, when epidemiological results are compared with COX-2/COX-1 ratios, there is a parallel relationship between gastrointestinal side effects and COX-2/COX-1 ratios.
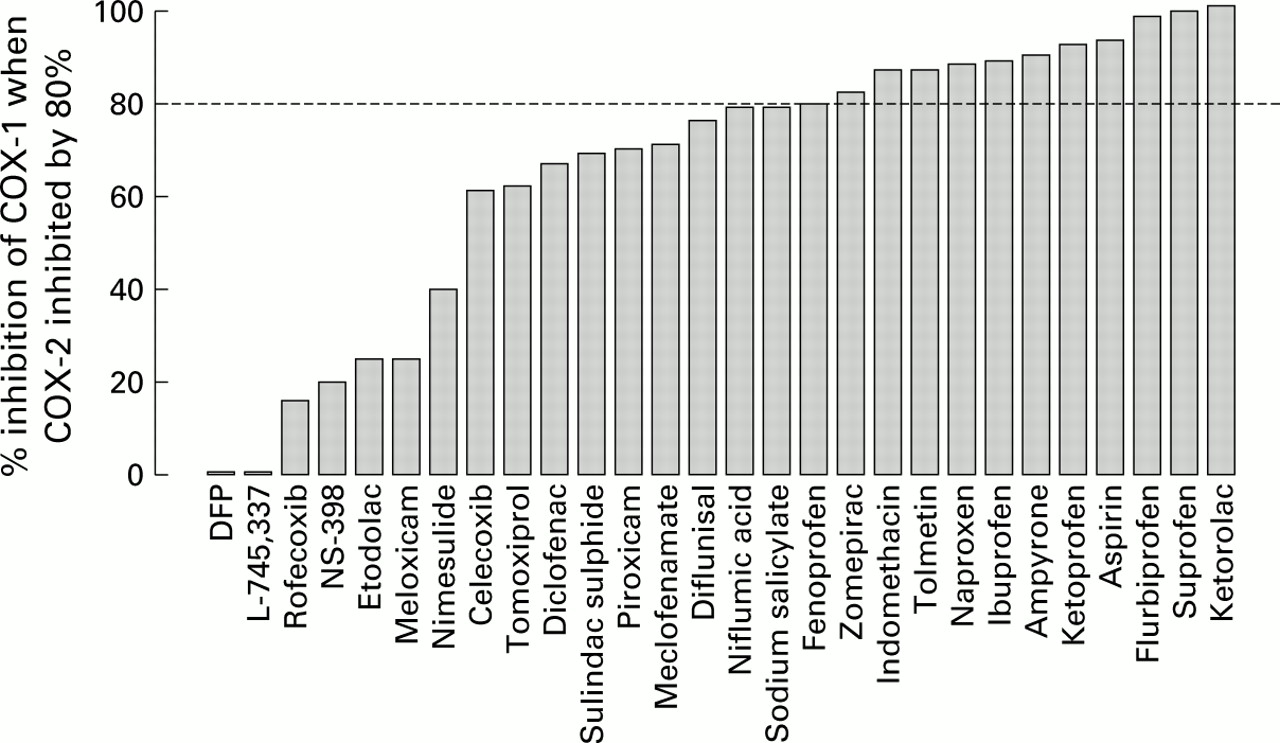
The conclusion that inhibition of COX-1 accounts for the gastric toxicity of NSAIDs is strongly reinforced by the work of Warneret al 69 in whole blood assays. They measured the effects of some 30 non-selective, selective, and highly selective COX-2 inhibitors in two types of whole blood assay for COX-1 and COX-2 (fig 1). There is substantial evidence that NSAIDs and COX-2 inhibitors have their maximum anti-inflammatory effect when COX-2 is inhibited by about 80%. Figure 2 shows the effects of these drugs on COX-1 at the 80% inhibitory concentration for COX-2. The NSAIDs that damage the stomach strongly inhibit COX-1.

Activity in modified whole blood assay of various NSAIDs on COX-1 at a dose that gives an 80% inhibition of COX-2. Those below the line have selectivity towards COX-1 and these are grouped with others that have a less than fivefold selectivity towards COX-2. The next group contains meloxicam, etodolac, celecoxib, and nimesulide which have 5–50 fold selectivity towards COX-2. Only rofecoxib has a greater than 50-fold selectivity for COX-2. Modified from Warner et al.69

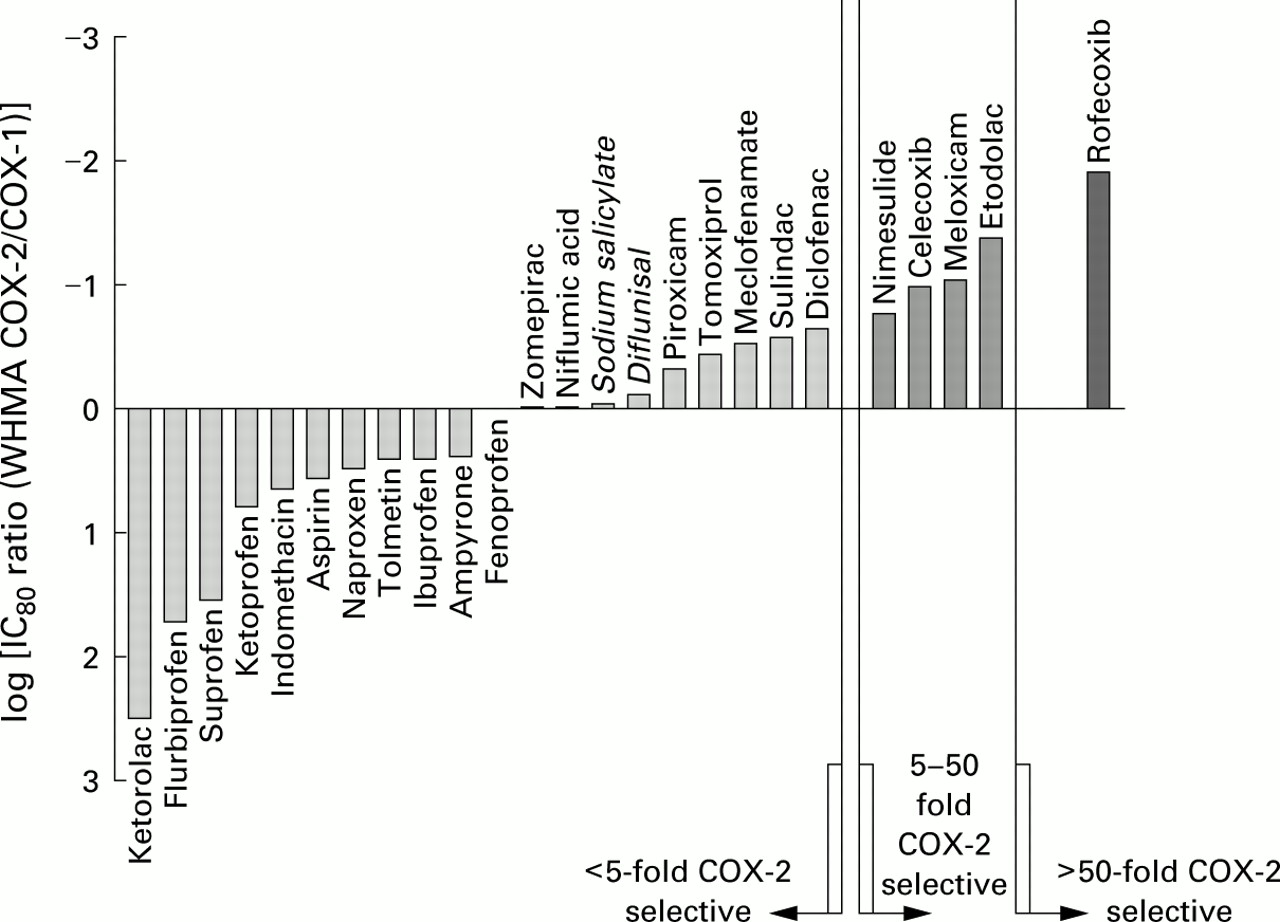{kind=link}
{kind=link}
Degree of inhibition of COX-1 in the whole blood assay plotted at concentrations of the drugs that give an inhibition of 80% of COX-2.
Selective inhibition of COX-2
Meloxicam, nimesulide, and etodolac were identified in the 1980s as potent anti-inflammatory drugs with low ulcerogenic activity in the rat stomach. In some instances this was also shown to parallel low activity against prostaglandin synthesis in the rat stomach. After the discovery of COX-2 these three drugs were each found to be selective COX-2 inhibitors.
Meloxicam is about 10 times more potent on COX-2 than on COX-1 in the human whole blood assay. It is marketed around the world for use in rheumatoid arthritis and osteoarthritis. In double blind trials in many thousands of patients with osteoarthritis meloxicam produced significantly fewer gastrointestinal adverse effects than the standard NSAIDs.71 ,72 Perforations, ulcerations, and bleedings occurred in fewer meloxicam treated patients than in those treated with piroxicam, diclofenac, or naproxen. The frequency of adverse events with meloxicam was significantly less than with piroxicam and naproxen (p<0.05).73 ,74
Etodolac is marketed for the treatment of osteoarthritis and rheumatoid arthritis. It is about 15 times more potent on COX-2 than on COX-1 in human whole blood. In healthy human volunteers etodolac twice daily did not suppress gastric mucosal prostaglandin production and caused less gastric damage than naproxen.75 Patients with osteoarthritis or rheumatoid arthritis obtained relief from symptoms with etodolac equal to other commonly used NSAIDs, but with a lower incidence of serious gastrointestinal toxicity.76
Nimesulide is about 10 times more active on COX-2 than on COX-1 in the human whole blood assay. In limited clinical trials in acute and chronic inflammation it was more effective than placebo or had comparable anti-inflammatory activity to established NSAIDs. Interestingly, nimesulide seems safe to use in aspirin sensitive asthmatic patients. Several recent studies in NSAID intolerant asthmatic patients have shown that therapeutic doses of nimesulide did not induce asthmatic attacks while high doses of 400 mg only precipitated mild asthma in 10% of patients.77 Perhaps aspirin induced asthma is associated with COX-1 inhibition?
Inhibitors designed to act on COX-2
Needleman and his group at Monsanto/Searle have made inhibitors that are some 1000 times more potent against COX-2 than against COX-1 in enzyme assays.78 One of these, SC-58635 (celecoxib), is an effective analgesic for moderate to severe pain following tooth extraction.79 Celecoxib given for seven days to human volunteers provided no evidence of gastric damage.80Interestingly, in our whole blood assay69 celecoxib is only 10 times more active against COX-2 than COX-1, which is a comparable selectivity to meloxicam.
Celecoxib was launched in December 1998 for the treatment of osteoarthritis and rheumatoid arthritis. Extensive clinical trial results have not yet been published. Interestingly, although doses up to 800 mg of celecoxib do not affect platelet aggregation in volunteers, there was an inhibition of serum thromboxane B2production of up to 70%, albeit with a shallow dose-response curve.35
Merck is also launching worldwide a COX-2 inhibitor, rofecoxib or Vioxx. In phase I studies a single dose of 250 mg daily for seven days (which is 10 times the anti-inflammatory dose) produced no adverse effects on the stomach mucosa, as evidenced by gastroscopy.81 After a single dose of 1 g there was no evidence of COX-1 inhibition in platelets, but activity of COX-2 in LPS stimulated monocytes ex vivo was reduced. For postoperative dental pain rofecoxib at 25–500 mg showed equal analgesic activity to ibuprofen82 and provided relief from symptoms in a six week study of osteoarthritis.83 Rofecoxib is also effective at 50 mg once daily in the pain of dysmenorrhoea.
Other possible therapeutic uses for selective COX-2 inhibitors
PREMATURE LABOUR
Prostaglandins are the cause of uterine contractions during labour. NSAIDs such as indomethacin will delay premature labour by inhibiting the production of prostaglandins, but will at the same time cause early closure of the ductus arteriosus and reduce urine production by the fetal kidneys.84 The delay in the birth process is probably the result of inhibition of COX-2 since mRNA for COX-2 increases substantially in the amnion and placenta immediately before and after the start of labour,58 whereas the side effects on the fetus are caused by inhibition of COX-1. One cause of preterm labour could be an intrauterine infection resulting in release of endogenous factors that increase prostaglandin production by upregulating COX-2.85 Nimesulide reduces prostaglandin synthesis in isolated fetal membranes and has been used successfully for a prolonged period to delay premature labour without manifesting the side effects of indomethacin on the fetus.84
COLON CANCER
Epidemiological studies have established a strong link between ingestion of aspirin and a reduced risk of developing colon cancer.86 ,87 Sulindac also caused reduction of prostaglandin synthesis and regression of adenomatous polyps in 11 of 15 patients with familial adenomatous polyposis (FAP), a condition in which many colorectal polyps develop spontaneously with eventual progression to tumours. Interestingly, COX-2 and not COX-1 is highly expressed in human and animal colon cancer cells as well as in human colorectal adenocarcinomas.45 ,46 In the mutantApc mouse, which is a model of FAP in humans, the spontaneous development of intestinal polyposis was strongly reduced either by deletion of the COX-2 gene or by treatment with a selective COX-2 inhibitor.88-90 Nimesulide also reduced the number and size of intestinal polyps inMin mice.91 The development of azoxymethane induced colon tumours over a year was inhibited in rats fed celecoxib.92 Thus, it is highly likely that COX-2 inhibitors could be used prophylactically to prevent colon cancer in genetically susceptible individuals without causing gastrointestinal damage themselves.
ALZHEIMER'S DISEASE
The connection between COX and Alzheimer's disease has been based mostly on epidemiology because of the lack of an animal model of the disease. A number of studies have shown a significantly reduced odds ratio for Alzheimer's disease in those taking NSAIDs as anti-inflammatory therapy.93-95 The Baltimore Longitudinal Study of Ageing96 with 1686 participants showed that the risk of developing Alzheimer's disease was reduced among users of NSAIDs, especially those who had taken the medications for two years or more. No decreased risk was evident with paracetamol or aspirin use. However, aspirin was probably taken in a dose too low to have an anti-inflammatory effect. The protective effect of NSAIDs is consistent with evidence of inflammatory activity in the pathophysiology of Alzheimer's disease. There is a strong interest in COX-2 in Alzheimer's disease, and Pasinetti and Aisen97have shown expression of COX-2 in the frontal cortex of the brain from these patients.
Conclusions
The identification of selective inhibitors of COX-2 will clearly provide important advances in the treatment of inflammation. Conventional NSAIDs lead to gastrointestinal side effects which include ulceration of the stomach, sometimes with subsequent perforation and deaths estimated at several thousand a year in the USA alone. The evidence is strong, both from animal tests and from the clinic, that selective COX-2 inhibitors will have greatly reduced side effects.
It is now becoming generally accepted that the unwanted side effects of NSAIDs result from their ability to inhibit COX-1 whilst their anti-inflammatory (therapeutic) effects result from inhibition of COX-2. This concept is now set in stone. Thus, selective COX-2 inhibitors will provide an important advance in anti-inflammatory treatment. In addition to their beneficial actions in inflammatory diseases, these drugs may be useful in the future for the prevention of colon cancer, Alzheimer's disease, or premature labour.
Finally, there are exciting clues to suggest that aspirin induced asthma may be a COX-1 dependent effect. If so, the selective COX-2 inhibitors may well avoid this distressing complication of treatment with NSAIDs.