Article Text

Statistics from Altmetric.com
Introduction
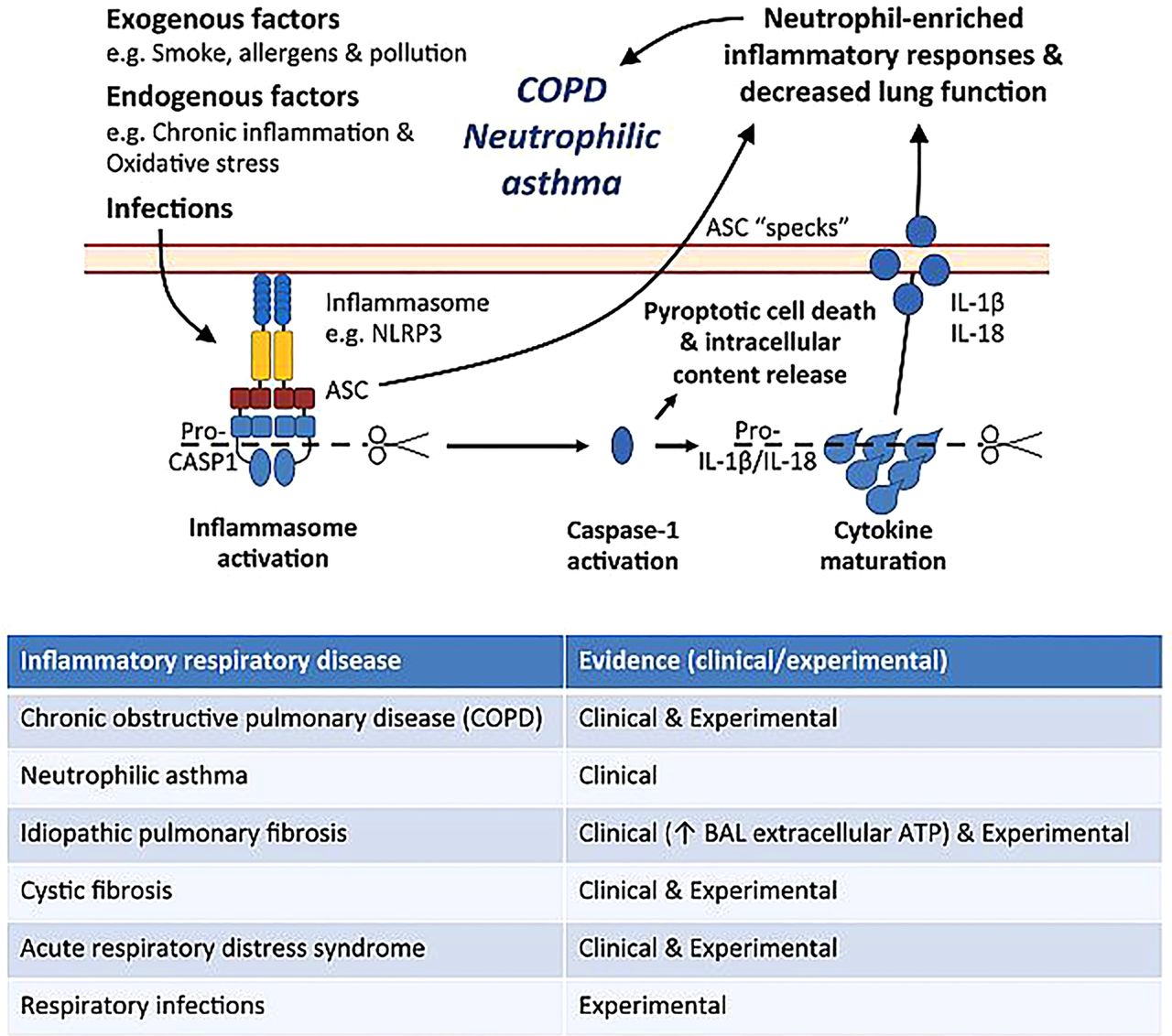
Inflammasomes have recently been identified to be critical and potent inducers of inflammation that when overactive may be targeted therapeutically in inflammatory diseases. Inflammasomes are multiprotein signalling complexes that control the maturation and release of pro-inflammatory cytokines in response to numerous exogenous, endogenous and pathogenic danger signals (figure 1).1 They mediate host responses, particularly interleukin (IL)-1β release and neutrophilic inflammatory responses, which are essential for protection against infection. However, recent evidence demonstrates that excessive inflammasome activation is a feature of numerous inflammatory diseases including COPD, neutrophilic asthma, idiopathic pulmonary fibrosis, cystic fibrosis, acute respiratory distress syndrome (ARDS) and respiratory infections (table in figure 1). Infections are the most widely recognised activators of inflammasomes; however, this field has recently been reviewed extensively and is beyond the scope of this article. Their roles in ARDS and potential for therapeutic targeting are well established.2 It is now emerging that inflammasomes are likely involved in the pathogenesis of COPD and asthma, and further investigation is urgently needed.
{kind=link}
Potential role for inflammasome activation in COPD and neutrophilic asthma. Numerous exogenous, endogenous and pathogenic danger signals (eg, ATP, monosodium urate crystals (MSU)) in the airways induce the assembly and activation of the inflammasome complex and synthesis of pro-interleukin (IL)-1β. Inflammasome activation by pathogen-associated molecular patterns (PAMPs) (eg, lipopolysaccharide (LPS) and peptidoglycan) results in proteolytic cleavage and activation of pro-caspase-1. Active caspase-1 in turn mediates the subsequent cleavage of pro-IL-1β that leads to the production and release of IL-1β and other pro-inflammatory mediators and cellular contents, including IL-18 and adaptor protein apoptosis-associated speck-like containing a caspase recruitment domain (ASC) specks. Excessive release of active IL-1β and ASC specks promotes neutrophil-dominated inflammatory responses in the airways that contribute to the pathogenesis of COPD and neutrophilic asthma. Complex interactions between inflammasomes, IL-1β, its receptors and intracellular signalling cascades that result in neutrophil-dominated inflammatory responses are involved in the pathogenesis of inflammasome-mediated, IL-1β-dependent respiratory diseases (see table). However, these events have been recently reviewed and are beyond the scope of this article. BAL, bronchoalveolar lavage.
To date, five main inflammasomes have been identified; NLRP1, NLRC4, RIG-I, AIM2 and NLRP3. They interact with other proteins termed the adaptor protein apoptosis-associated speck-like containing a caspase recruitment domain (ASC) and pro-caspase-1 to form distinct inflammasome complexes (figure 1). The expression, complex formation and activation of inflammasomes are initiated by danger signals that are activated by bacterial components (pathogen-associated molecular patterns (PAMPs)) and/or host-derived factors (damage-associated molecular patterns (DAMPs)) that are released during infection, injury, inflammation or stress. Inflammasome activation initiates a cascade of events that result in the recruitment and cleavage of pro-caspase-1 into active caspase-1 molecules that then mediate the cleavage of pro-IL-1β into its active form, IL-1β, within the activated cell. Cleavage of pro-IL-18 into active IL-18 also occurs. Caspase-1 also triggers an uncontrolled form of cell death known as pyroptosis, which results in the release of pro-inflammatory mediators and other cellular contents into the extracellular environment, further inducing inflammatory responses. Therefore, activation of the inflammasome is crucial for the induction of caspase-1-dependent processing, maturation and secretion of IL-1β. This highlights a key role for the inflammasome in IL-1β-mediated immune responses (figure 1).1 There are also non-inflammasome-mediated mechanisms that activate and modify IL-1β and IL-18 responses including caspase-8 and granzyme B, and numerous serine proteases and tryptases. However, a discussion of these is outside the scope of this review.
Of the inflammasomes, the properties and functions of NLRP3 are best characterised.1 NLRP3 expression, assembly and the production of pro-IL-1β are induced by a range of pro-inflammatory signalling events, such as those triggered by families of receptors including the Toll-like receptors that recognise PAMPs such as lipopolysaccharide and peptidoglycan.1 Although inflammasome-dependent caspase-1 activation can be observed in the absence of PAMPs, the resulting levels of active IL-1β are minimal.1 Furthermore, the activation of the assembled NLRP3 complex, and subsequent generation of caspase-1, requires the presence of DAMPs such as extracellular ATP and monosodium urate crystals, which are produced by cells under inflammatory conditions.1 Therefore, the maximal production of mature IL-1β requires at least two distinct signals, the first is a pro-inflammatory PAMP stimulus that induces the expression of pro-IL-1β and inflammasome components and the second is DAMP-induced inflammasome complex formation and activation and caspase-1-mediated maturation of pro-IL-1β and release of active IL-1β (figure 1).1 See Schroder and Tschopp1 for a comprehensive discussion of the mechanisms of inflammasome activation.
Cigarette smoke not only contains noxious chemicals but also substantial amounts of microbial components and induces tissue damage in the airways,3 which likely contribute to the pathogenesis of COPD and neutrophilic asthma in smokers (figure 1). Indeed, since microbial PAMPs and DAMPs are the essential stimuli for inflammasome activity cigarette smoke exposure alone may be sufficient for activity and the increased production of IL-1β. Furthermore, both COPD and severe, steroid-insensitive forms of neutrophilic asthma have been associated with viral and bacterial respiratory infections, particularly during exacerbations of disease. This brief review focuses on recent studies that highlight potential roles for inflammasome activation in COPD and neutrophilic asthma.
Inflammasomes in COPD
Studies have found increased inflammasome activation and IL-1β in the lungs of patients with COPD and neutrophilic asthma3–5 and also in experimental models.6 ,7 Indeed, we, and others, demonstrated that IL-1β expression is increased during the development of cigarette smoke-induced experimental COPD6 ,7 and have since shown increased protein production (unpublished data). A recent study by Franklin et al3 highlights a role for inflammasome activation in the pathogenesis of COPD as evidenced by the accumulation of ASC ‘specks’ in the lungs of patients with COPD and in murine models of the disease. The ASC protein is a key component of inflammasomes and is required for the recruitment of pro-caspase-1 to the inflammasome complex and the production of caspase-1. However, during pyroptotic cell death that follows inflammasome activation, ASC specks are released from activated cells and accumulate in extracellular spaces.3 These ASC specks retain their ability to mature pro-IL-1β in the extracellular environment and, importantly, are readily phagocytosed by macrophages and promote the production of IL-1β by these cells.3 The authors also showed that ASC specks were capable of recruiting neutrophils and inflammatory monocytes when injected into mice. These findings demonstrate that ASC specks have the potential to perpetuate inflammatory responses, following inflammasome activation. Importantly, the authors also show that bronchoalveolar lavage (BAL) collected from patients with COPD and a cigarette smoke-induced murine model of experimental COPD were highly enriched with extracellular ASC specks compared with BAL from healthy controls. This and several other studies indicate that inflammasome activation, and subsequent ASC speck accumulation, may play important roles in inflammatory processes in COPD. However, further investigations are required to confirm a mechanistic involvement.
In contrast, a recent article published in Thorax found no correlation between NLRP3, caspase-1 and IL-1β responses and the severity of COPD.8 The authors examined the expression of several innate and NLRP3 inflammasome-associated factors in bronchial mucosa and BAL from patients with stable COPD of different severity and controls. While they did show a small increase in NLRP3 expression in the epithelium of patients with severe compared with mild COPD, they could not detect IL-1β and caspase-1 responses in samples from patients with COPD or non-smokers and smokers without COPD.8 The inflammasome inhibitory molecules NALP7 and IL-37 were increased in patients with COPD compared with control smokers. They note that their observations are in contrast to findings in several other studies, which demonstrated increased IL-1β in the airways and serum of patients with COPD compared with controls. Furthermore, while their findings suggested that NLRP3-mediated, IL-1β responses play a limited role in patients with stable COPD, they are important in respiratory infections.8 Therefore, inflammasomes may be important in infection-associated exacerbations of the disease. Whether they are important in COPD development has not been investigated.
Inflammasomes in neutrophilic asthma
Much recent work has demonstrated that asthma is a heterogeneous disease. While some patients exhibit a T-helper lymphocyte type (Th)2-mediated, eosinophilic inflammatory endotype, recent clinical studies have shown that moderate-to-severe forms of asthma can be associated with increased Th1- and/or Th17- responses and non-eosinophilic subtypes of disease that are characterised by predominantly monocytic or neutrophilic, rather than eosinophilic, airway inflammation. The identification of different subtypes of asthma highlights the potential for developing better therapies that target the specific factors that drive individual disease phenotypes. In particular, improved therapeutic strategies are required for neutrophilic asthmatics as these patients typically have more severe disease and are relatively insensitive to steroid treatment and, therefore, have limited treatment options. The development of such tailored therapeutic strategies for neutrophilic asthma has been hampered by a lack of understanding of the mechanisms that lead to disease. Two recent studies have shown that inflammasome-mediated IL-1β responses may play a role in the pathogenesis of neutrophilic asthma.
Baines et al investigated asthma phenotypes at a transcriptional level using gene expression profiling of induced sputum. They showed that asthmatics with a neutrophil-dominated inflammatory profile had a distinct sputum gene expression profile to that of asthmatics with eosinophil-dominated and/or macrophage-dominated disease.4 Genes associated with the IL-1β signalling pathway (eg, IL1B, IRAK2, IRAK3, IL1R2) were significantly upregulated in neutrophilic asthma.4 A subsequent study by Simpson et al provided strong evidence that these increased IL-1β responses in neutrophilic compared with other forms of asthma may be driven by increased NLRP3 inflammasome activation.5 They showed that neutrophilic asthmatics had significantly higher levels of NLRP3, caspase-1 and IL-1β in their sputum than eosinophilic and paucigranulocytic asthmatics.5 Furthermore, NLRP3 and caspase-1 proteins were enriched in macrophages and neutrophils in the sputum of neutrophilic, but not eosinophilic or paucigranulocytic, asthma. Many neutrophilic asthmatics, including those in the two studies described, are allergic/atopic and aberrant responses to allergens are still a pathogenic feature of this form of the disease. Together, these studies highlight associations between neutrophilic asthma and increased IL-1β responses that may be driven by excessive NLRP3 inflammasome activation. This raises the possibility of targeting the inflammasome and/or IL-1β as a therapeutic strategy in neutrophil-dominated forms of severe, steroid-insensitive asthma.
Although poorly understood, what is known of the mechanistic roles of IL-1β, inflammasomes and neutrophilic inflammation in COPD and neutrophilic asthma are described in the studies highlighted above.
Conclusions
Inflammasomes play key roles in host defence against infection. Their activation is also increased in chronic inflammatory diseases. Recent studies show associations between increased inflammasome activation and IL-1β responses in COPD and neutrophilic asthma. However, the relative contribution of inflammasomes in inducing IL-1β responses and the pathological roles played by inflammasome-mediated IL-1β in these diseases remain to be elucidated. Furthermore, the underlying contributors to inflammasome activation in these conditions are yet to be fully explored. Inflammasome-mediated IL-1β responses are induced in the lung during smoke exposure and infection. Thus, it is likely that excessive inflammasome activation and IL-1β responses contribute to the complex inflammatory processes that underpin the development of COPD and neutrophilic asthma as well as infection-associated exacerbations that promote progression into more severe disease (figure 1). This probably occurs in combination with other disease drivers such as oxidative stress, epigenetics and genetics. The articles we highlight demonstrate that comprehensive studies that increase the understanding of the nature of the association between inflammasome-mediated IL-1β responses and the aetiological drivers of inflammasome activation in COPD and neutrophilic asthma are required. Such studies would inform the development of improved therapies that target inflammasomes for the treatment of these diseases. Tools that allow the dissection of the specific roles of inflammasomes in combination with inhibitors of downstream components of inflammasome-mediated inflammation and representative animal models of COPD and severe, steroid-insensitive, neutrophilic asthma are required to enable such studies.
Footnotes
Contributors All authors contributed to the conception of the review, the literature search, the manuscript drafting and its revision. All authors approve the version to be published and are accountable for the accuracy and integrity of the work.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.