Article Text

Abstract
Background Pulmonary hypertension in idiopathic pulmonary fibrosis (IPF) is indicative of a poor prognosis. Recent evidence suggests that tetrahydrobiopterin (BH4), the cofactor of nitric oxide synthase (NOS), is involved in pulmonary hypertension and that pulmonary artery endothelial-to-mesenchymal transition (EnMT) may contribute to pulmonary fibrosis. However, the role of BH4 in pulmonary remodelling secondary to pulmonary fibrosis is unknown. This study examined the BH4 system in plasma and pulmonary arteries from patients with IPF as well as the antiremodelling and antifibrotic effects of the BH4 precursor sepiapterin in rat bleomycin-induced pulmonary fibrosis and in vitro EnMT models.
Methods BH4 and nitrotyrosine were measured by high-performance liquid chromatography and ELISA, respectively. Expression of sepiapterin reductase (SPR), GTP cyclohydrolase 1 (GCH-1), endothelial NOS (eNOS) and inducible NOS (iNOS) were measured by quantitative PCR and immunohistochemistry.
Results BH4 plasma levels were downregulated in patients with IPF compared with controls while nitrites, nitrates and nitrotyrosine were upregulated. GCH-1 and eNOS were absent in pulmonary arteries of patients with IPF; however, iNOS expression increased while SPR expression was unchanged. In rats, oral sepiapterin (10 mg/kg twice daily) attenuated bleomycin-induced pulmonary fibrosis, mortality, vascular remodelling and pulmonary hypertension by increasing rat plasma BH4, decreasing plasma nitrotyrosine and increasing vascular eNOS and GCH-1 expression. Both transforming growth factor β1 and endothelin-1 induced EnMT by decreasing BH4 and eNOS expression. In vitro administration of sepiapterin increased endothelial BH4 and inhibited EnMT in human pulmonary artery endothelial cells.
Conclusions Targeting the BH4 synthesis ‘salvage pathway’ with sepiapterin may be a new therapeutic strategy to attenuate pulmonary hypertension in IPF.
- Idiopathic pulmonary fibrosis
- Oxidative Stress
- Interstitial Fibrosis
Statistics from Altmetric.com
Key messages
What is the key question?
-
Could targeting the tetrahydrobiopterin system attenuate pulmonary artery remodelling in pulmonary fibrosis?
What is the bottom line?
-
Tetrahydrobiopterin (BH4) is downregulated in plasma and pulmonary arteries from patients with idiopathic pulmonary fibrosis. Stimulation of the BH4 biosynthesis ‘salvage pathway’ by oral administration of sepiapterin reduced pulmonary remodelling and pulmonary fibrosis in a bleomycin-induced pulmonary fibrosis animal model and inhibited the endothelial-to-mesenchymal transition as a source of myofibroblasts in human pulmonary artery endothelial cells.
Why read on?
-
The results suggest that targeting the BH4 synthesis ‘salvage pathway’ may be a new therapeutic strategy to attenuate pulmonary hypertension in idiopathic pulmonary fibrosis.
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common idiopathic interstitial pulmonary disease. It is a fatal disorder with a median survival of 2.5–5 years as no effective treatment exists. Pulmonary hypertension is recognised as a severe complication of IPF associated with poor survival.1 Recent epidemiology studies have reported a prevalence of pulmonary hypertension in IPF of 32–84% depending on the stage of IPF progression.1 The pathogenesis of pulmonary hypertension in IPF reflects a multifactorial and complex process involving a variety of pathways and mediators. In the early stages, vascular disease is affected by muscularisation followed by fibrous vascular atrophy and pronounced intimal fibrosis. Subsequently, there is fibrous atrophy of the media and, finally, fibrous ablation of affected vessels. Perivascular fibrosis may influence the distension of the pulmonary vessels, possibly augmenting pulmonary vascular resistance and pulmonary artery pressure.2 The blood vessels may themselves participate in the genesis of IPF, and similar cytokine disruptions have been described in IPF and pulmonary hypertension including changes in levels of transforming growth factor β (TGF-β), connective tissue growth factor (CTGF) or endothelin-1 (ET-1). This suggests that these disorders share pathogenic features and that one may influence, generate or perpetuate the other.
The various cellular processes involved in pulmonary artery remodelling are observed in pulmonary hypertension-associated IPF, including endothelial dysfunction,3 endothelial-to-mesenchymal transition as a source of fibroblasts,4 as well as fibroblast and pulmonary artery smooth muscle proliferation and transition to myofibroblasts.5 Dysfunction of pulmonary artery endothelial cells is present in IPF3 and may manifest as enhanced synthesis of ET-1, TGF-β, reactive oxygen/nitrogen species and platelet-derived growth factor6 or decreased synthesis of vasodilators and anti-smooth muscle cell proliferation molecules such as nitric oxide (NO) or prostacyclin.7
NO is synthesised by three distinct nitric oxide synthases (NOS), two of which are expressed constitutively in neurons (nNOS) and vascular endothelial cells (eNOS). The expression of a third isoform (iNOS) is induced by a number of cytokines in a broad spectrum of cell types.8 NO bioavailability is reduced in pulmonary hypertension due to reduced NO synthesis or increased NO consumption by reactive oxygen species. All three NOS isoforms additionally require tetrahydrobiopterin (BH4) for their catalytic activity.8 Thus, when BH4 levels are adequate, NOS produces NO; low levels of BH4 cause uncoupled NOS enzymatic activity, generating NO, superoxide and peroxynitrite, thus contributing to vascular oxidative stress and endothelial dysfunction. BH4 bioavailability is determined by the balance between de novo synthesis and oxidative degradation to non-active BH2. GTP cyclohydrolase 1 is the first step and rate-limiting enzyme for de novo BH4 biosynthesis. An alternative ‘salvage pathway’ transforms exogenous sepiapterin to BH4 through the enzyme sepiapterin reductase.9
Pulmonary artery expression of eNOS is decreased or absent in the endothelium of patients with IPF while iNOS is highly expressed, in parallel with an increase in reactive oxygen species and nitrotyrosine, in the lung tissue of these patients, which suggests uncoupled NOS activity.10 Recent studies have shown that BH4 or its precursor sepiapterin possesses antioxidant and antifibrotic properties in cardiovascular diseases.11 ,12 However, the role of the BH4 system in IPF, as well as the associated pulmonary hypertension, is currently unknown. Based on these observations, we hypothesise that: (1) levels of BH4 in patients with IPF are downregulated by alteration of the enzymes required for BH4 biosynthesis, thus increasing uncoupled NOS and oxidative stress; (2) oral administration of sepiapterin as a source of intracellular BH4 may improve pulmonary hypertension and lung fibrosis in a model of bleomycin-induced lung fibrosis; and (3) endothelial-to-mesenchymal transition as a source of pulmonary fibroblasts is inhibited by sepiapterin in vitro in cultured pulmonary artery endothelial cells.
Methods
Details of the methods are shown in the online supplement.
Patients with IPF
Fibrotic lung samples were obtained at lung transplantation surgery or by open lung biopsy for histological diagnosis of the disease. Age-matched normal control lungs were collected from a non-involved segment remote from the solitary lesion of patients undergoing thoracic surgery for removal of a primary lung tumour.
Animal model
Animal studies were performed in accordance with the guidelines of the Committee of Animal Ethics and Well-being of the University of Valencia. Wistar rats were instilled on day 1 with a single intratracheal dose of 3.75 U/kg bleomycin. Sham-treated rats received an identical volume of intratracheal saline instead of bleomycin. Twice daily oral doses of sepiapterin (10 mg/kg) were administered via an intra-oesophageal cannula from day 1 to 21. At the end of the treatment period (day 21) the rats were killed. Right ventricular hypertrophy was measured as described previously.13 Right ventricular systolic pressure (RVSP) was determined by right heart catheterisation.
Histological, immunohistochemical and immunofluorescence studies
The severity of lung fibrosis was scored on a scale from 0 (normal lung) to 8 (total fibrotic obliteration of fields) according to the Ashcroft scoring system.14 Pulmonary artery wall thickness was determined as reported previously.15 For immunohistochemical analysis of rat and human lungs, sections were immunostained with α-smooth muscle actin, sepiapterin reductase, GTP cyclohydrolase 1, eNOS and iNOS antibodies. Staining intensities of the various antibodies were scored and summed to obtain a composite score of 0–12, as described previously.16 Immunofluorescence of CD31, collagen type I, α-smooth muscle actin and vascular endothelial (VE)-cadherin was assessed in human pulmonary artery endothelial cells.
Determination of BH4/BH2, nitrites and nitrotyrosine
BH4 and BH2 in plasma, pulmonary artery tissue and cell culture supernatant were measured by high-performance liquid chromatography. NO and nitrotyrosine concentrations were determined using a commercially available ELISA (see online supplement for details).
Isolation of human pulmonary artery endothelial cells
Cell culture experiments were performed using human pulmonary artery endothelial cells isolated from the pulmonary arteries of normal lungs using a commercially available Dynabeads CD31 endothelial cell kit (Dynal Biotech, Germany).
Quantitative PCR and western blotting
Transcript levels were quantified using the 2−ΔΔCt method. Western blotting was used to detect changes in TGF-β1, ET-1 and phosphorylated Smad3 (p-Smad3) levels in lung tissue and human pulmonary artery endothelial cells.
DCF fluorescence measurement of reactive oxygen species
H2DCF-DA (2′,7′-dichlorodihydrofluorescein diacetate; Molecular Probes, Nottingham, UK) was used to monitor intracellular reactive oxygen species in human pulmonary artery endothelial cells.
Statistical analysis
Statistical analysis was carried out by parametric (animal and cell culture studies) or non-parametric (human studies) analysis, as appropriate; p<0.05 was considered to indicate statistical significance. For non-parametric tests, data are displayed as medians, IQR and minimum and maximum values analysed by the Mann–Whitney test. Parametric data are expressed as means±SE of n experiments using the Student t test or one-way or two-way analysis of variance followed by the Bonferroni post hoc test.
Results
Bioavailability of BH4 in patients with IPF
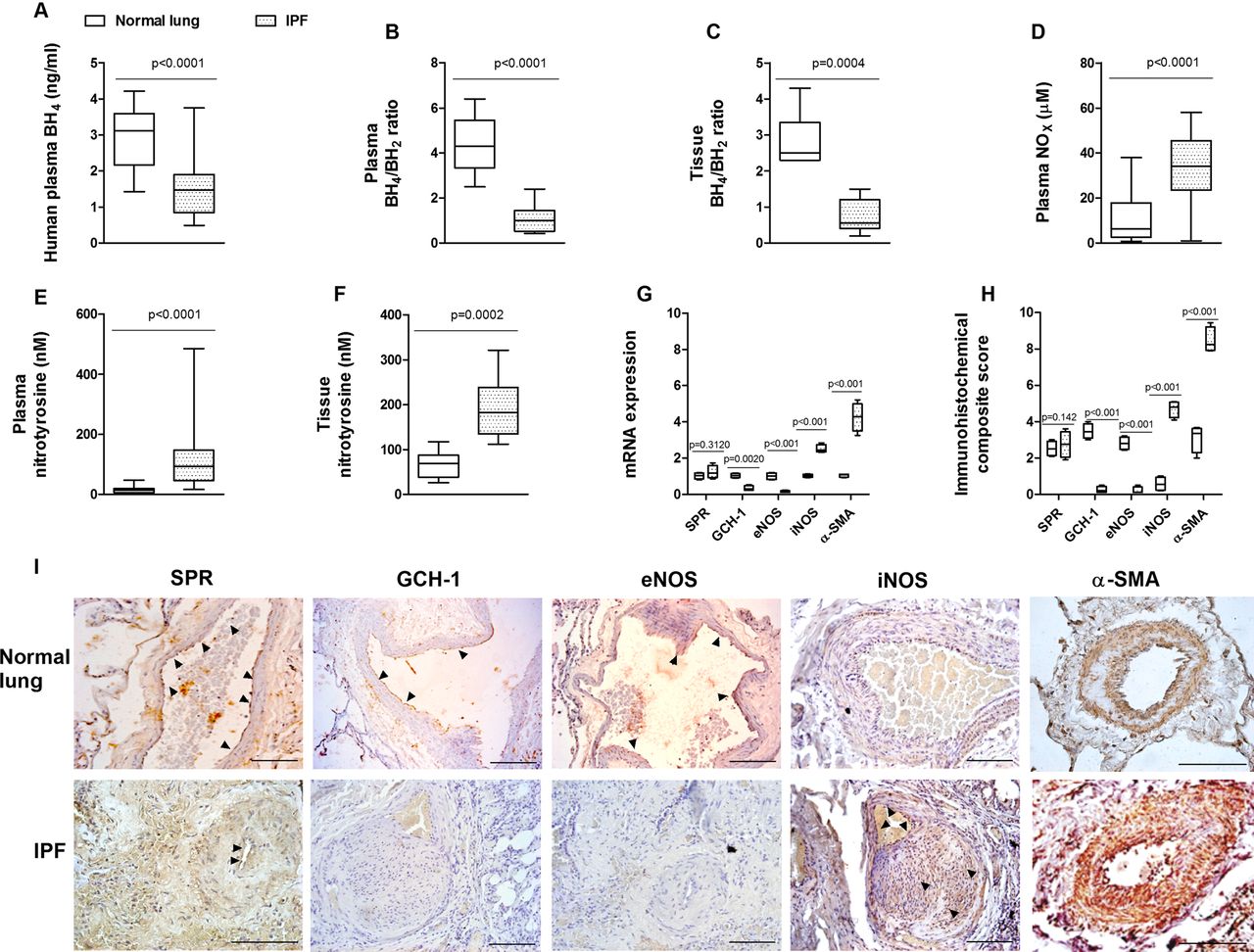
Both controls and patients with IPF were prospectively recruited from 2008 to 2012. Clinical data from enrolled patients are shown in table 1. Levels of BH4 were significantly downregulated in plasma from patients with IPF (1.46±0.69 ng/mL) compared with healthy controls (2.93±0.8 ng/mL; figure 1A). Additionally, BH4/BH2 was lower in plasma and pulmonary artery tissue from patients with IPF (figure 1B,C). Plasma levels of NOx (nitrate and nitrite) were increased in patients with IPF compared with healthy subjects (figure 1D). Increased levels of plasma and pulmonary artery tissue nitrotyrosine were also observed in patients with IPF compared with healthy subjects (figure 1E,F). In contrast to control normal lungs, pulmonary arteries from patients with IPF showed a lack of gene and protein expression of GTP cyclohydrolase 1 and eNOS, while iNOS and α-smooth muscle actin expression was upregulated in pulmonary arteries (figure 1G–I). However, the expression of sepiapterin reductase was similar in pulmonary arteries from control subjects and patients with IPF. No correlation between clinical data or pulmonary hypertension with BH4 values was identified in patients with IPF.
Clinical characteristics

Tetrahydrobiopterin (BH4) bioavailability in idiopathic pulmonary fibrosis (IPF). (A) Plasma BH4 levels, (B) plasma BH4/dihydrobiopterin (BH2) levels, (C) pulmonary artery tissue BH4/BH2 levels, (D) plasma nitrites+nitrates (NOx), (E) plasma nitrotyrosine, (F) pulmonary artery tissue levels of BH4 in healthy subjects (n=30) and patients with IPF (n=36), (G) expression of sepiapterin reductase (SPR), GTP cyclohydrolase 1 (GCH-1), endothelial nitric oxide synthase (eNOS), inducible nitric oxide synthase (iNOS) and α-smooth muscle actin (α-SMA) genes in pulmonary arteries from 21 normal controls and 17 patients with IPF. Data are expressed as ratios to GAPDH mRNA levels. The box plot in (F) represents the composite score of SPR, GCH-1, eNOS, iNOS and α-SMA markers across pulmonary arteries in 10 slices per patient. Data are presented as a box and whisker plot of medians, IQR and minimum and maximum values. p Values were obtained by Mann–Whitney test. (H, I) Immunohistochemistry of pulmonary arteries. Pulmonary artery sections from normal lungs (n=21) and patients with IPF (n=17) were immunostained for SPR, GCH-1, eNOS, iNOS and α-SMA (brown) and counterstained with haematoxylin. Black arrows show positively immunostained regions. Representative immunohistochemistry images are shown. Scale bar=100 µm. The IgG isotype control was negative.
Effects of sepiapterin on bleomycin-induced lung fibrosis and mortality
Following 21 days of intratracheal bleomycin instillation, a marked loss of body weight and an increase in lung mass and mortality were observed (figure 2A–C). Twice daily oral administration of sepiapterin (10 mg/kg) reduced body weight loss and increased lung mass secondary to fibrosis, and increased survival. Bleomycin induced a fibrotic response in the lung, with enhanced expression of fibrotic markers TGF-β1, ET-1, CTGF and collagen type I (figure 2D) and increased deposition of collagen, as visualised by Masson's trichrome staining (figure 2E). Sepiapterin alleviated histologically observed multifocal fibrotic lesions, resulting in fewer organised and smaller foci and reduced septal enlargement with a diminished Ashcroft fibrosis score (figure 2F).
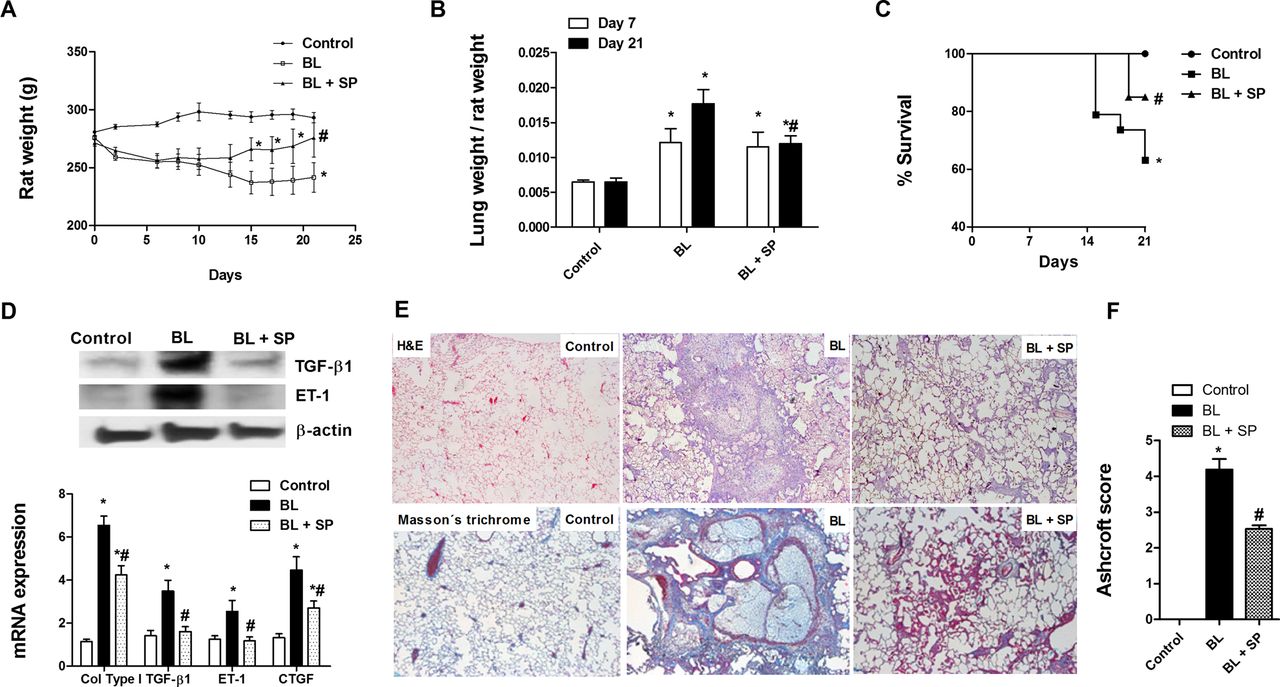
Sepiapterin improves bleomycin-induced pulmonary fibrosis. Wistar rats received a single intratracheal dose of bleomycin (BL; 3.75 U/kg) on day 1. Sepiapterin (SP; 10 mg/kg twice daily orally) or vehicle was administered from day 1 until analysis at day 21 (n=10 per group). (A) Rat weight and (B) lung mass were monitored for 21 days. (C) Kaplan–Meier survival curve showing 60% survival over the 21-day BL period (square), 83% in the SP-treated group (triangle) and 100% in the control rat group (circle); *p<0.05, log-rank test. (D) Total lung protein and gene expression of the profibrotic markers transforming growth factor β1 (TGF-β1), endothelin 1 (ET-1), collagen type I (col type I) and connective tissue growth factor (CTGF). Data are expressed as ratios to GAPDH mRNA levels and to β-actin protein levels, normalised to the control group. (E) Representative western blot images of total TGF-β1 and ET-1; H&E staining (upper panels, 40×) and Masson's trichrome (lower panels, 40×; collagen stained in blue) of controls, BL and BL+SP. (F) Fibrosis Ashcroft scores were assessed as described in the Methods section. Results are expressed as means±SE (n=10). Statistical significance was assessed using a t test or one-way analysis of variance followed by a Bonferroni post hoc test. *p<0.05 vs control, #p<0.05 vs BL.
Sepiapterin improves bleomycin-induced pulmonary remodelling
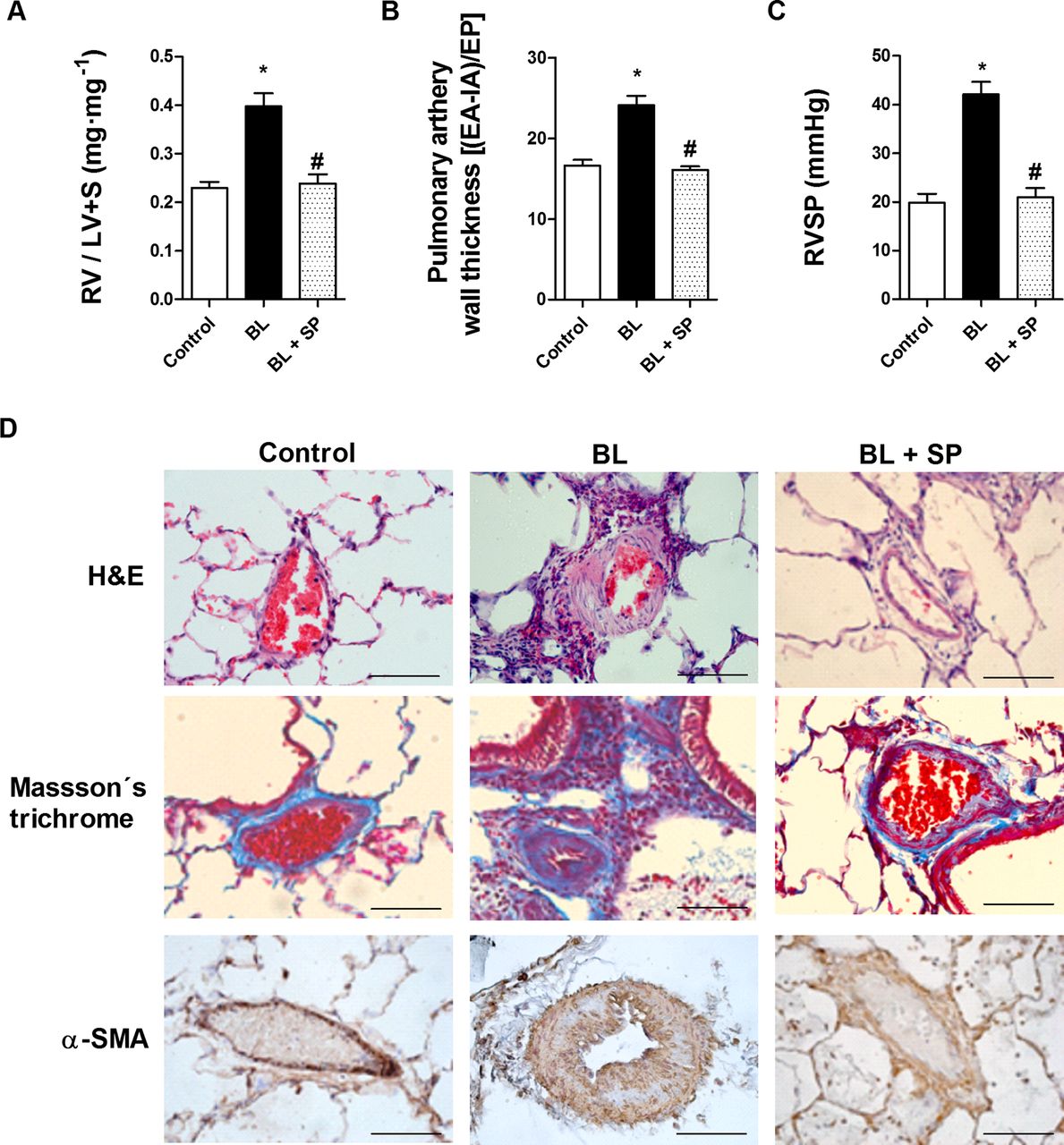
Right ventricular hypertrophy (RV/LV+septum) and pulmonary vascular remodelling developed following bleomycin treatment (figure 3A,B). By day 21, pulmonary hypertension was observed in the bleomycin group, as peak RVSP had increased from 19.8±4.2 mm Hg in control rats to 42.12±5.1 mm Hg in the bleomycin group (figure 3C). Sepiapterin suppressed RV hypertrophy almost completely, pulmonary vascular remodelling and normalised pulmonary hypertension to 20.9±4.7 mm Hg at day 21. Sepiapterin also reduced collagen deposition visualised by Masson's trichrome staining and muscularisation visualised by α-smooth muscle actin immunostaining in the walls of the pulmonary arteries (figure 3D).

Analysis of bleomycin-induced pulmonary vascular remodelling and right ventricular hypertrophy. Rats received a single intratracheal dose of bleomycin (BL; 3.75 U/kg) on day 1. Sepiapterin (SP; 10 mg/kg twice daily orally) or vehicle was administered from day 1 until analysis at day 21 (n=10 per group). (A) Right ventricular hypertrophy (expressed as RV/LV+S ratio). (B) Pulmonary artery wall thickness was calculated by dividing the wall area (EA-IA) by the external perimeter (EP) in intra-acinar pulmonary arteries immunostained with α-smooth muscle actin (α-SMA) antibody. (C) Right ventricular systolic pressure (RVSP; mm Hg) in control rats, BL and BL+SP 21 days after BL instillation. (D) H&E staining (upper panels, scale bar=20 µm) and Masson's trichrome (lower panels, collagen stained in blue) of controls, BL and BL+SP. Results are expressed as means±SE (n=10). Statistical significance was assessed using one-way analysis of variance followed by a Bonferroni post hoc test. *p<0.05 vs control, #p<0.05 vs BL.
Effects of sepiapterin on BH4 and NO bioavailability in bleomycin-treated rats
Absolute plasma levels of BH4 at day 21 were significantly downregulated in the bleomycin group (figure 4A). Decreased levels of BH4 were accompanied by an increase in partially oxidised BH2 levels, so the plasma BH4/BH2 ratio was decreased in the bleomycin group (figure 4B). In plasma from untreated rats the NOx concentration was significantly lower than in bleomycin-treated rats (control 9.2±2.5 μM vs bleomycin 19.5±5.3 μM; figure 4C). Additionally, plasma levels of nitrotyrosine were increased approximately twofold over control rats (figure 4D). Oral administration of sepiapterin increased plasma BH4 as well as BH4/BH2 to levels similar to control rats (figure 4A,B). Sepiapterin inhibited bleomycin-induced upregulation of plasma NOx and nitrotyrosine to levels similar to those of control rats (figure 4C,D). Immunohistochemical analysis of pulmonary arteries showed a significant decrease in GTP cyclohydrolase 1 expression (figure 4E,F), but no change in sepiapterin reductase, after 21 days of bleomycin exposure. In control rats, eNOS expression was located primarily in endothelial cells while, in the bleomycin group, eNOS expression was not observed. In contrast, iNOS was highly induced in pulmonary arteries of bleomycin-treated rats. Oral administration of the sepiapterin reductase substrate sepiapterin to bleomycin-treated rats significantly increased GTP cyclohydrolase 1 and eNOS expression and partially inhibited iNOS induction in pulmonary arteries (figure 4E,F).
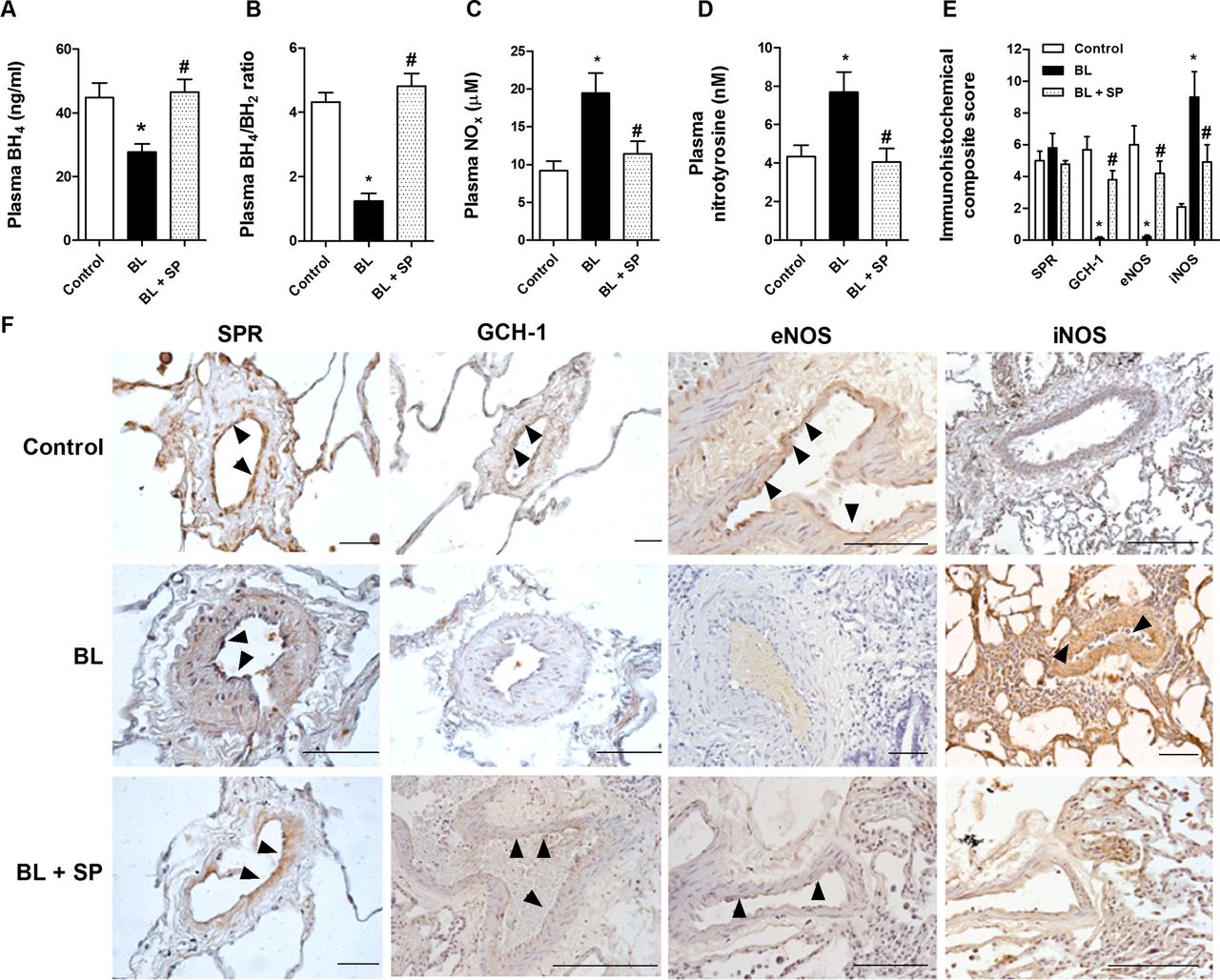
Sepiapterin increases tetrahydrobiopterin (BH4) and reduces nitrotyrosine plasma levels in bleomycin-treated rats. Rats received a single intratracheal dose of bleomycin (BL; 3.75 U/kg) on day 1. Sepiapterin (SP; 10 mg/kg twice daily orally) or vehicle was administered from day 1 until analysis at day 21 (n=10 per group). (A) BH4 levels, (B) BH4/dihydrobiopterin (BH2), (C) nitrites+nitrates (NOx), (D) nitrotyrosine rat plasma levels (n=10 per group). (E, F) Immunohistochemistry of pulmonary arteries from control vehicle, BL and BL+SP groups were immunostained for sepiapterin reductase (SPR), GTP cyclohydrolase 1 (GCH-1), endothelial nitric oxide synthase (eNOS) and inducible nitric oxide synthase (iNOS) (brown) and counterstained with haematoxylin. Black arrows show positively immunostained regions. Representative immunohistochemistry images are shown. Scale bar=50 µm. The IgG isotype control was negative. The box plot in (E) represents the composite score of SPR, GCH-1, eNOS and iNOS markers across the pulmonary arteries in 10 slices per animal. Results are expressed as means±SE (n=10). Statistical significance was assessed using one-way analysis of variance followed by a Bonferroni post hoc test. *p<0.05 vs control, #p<0.05 vs BL.
Sepiapterin inhibits the endothelial-to-mesenchymal transition induced by fibrotic mediators
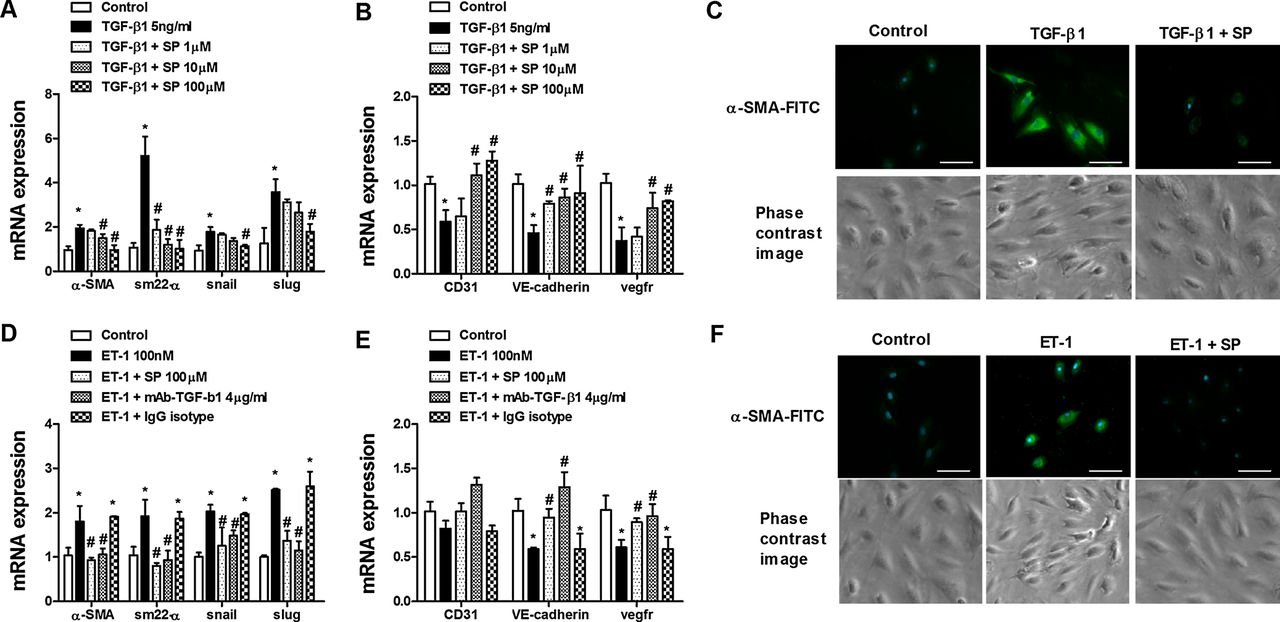
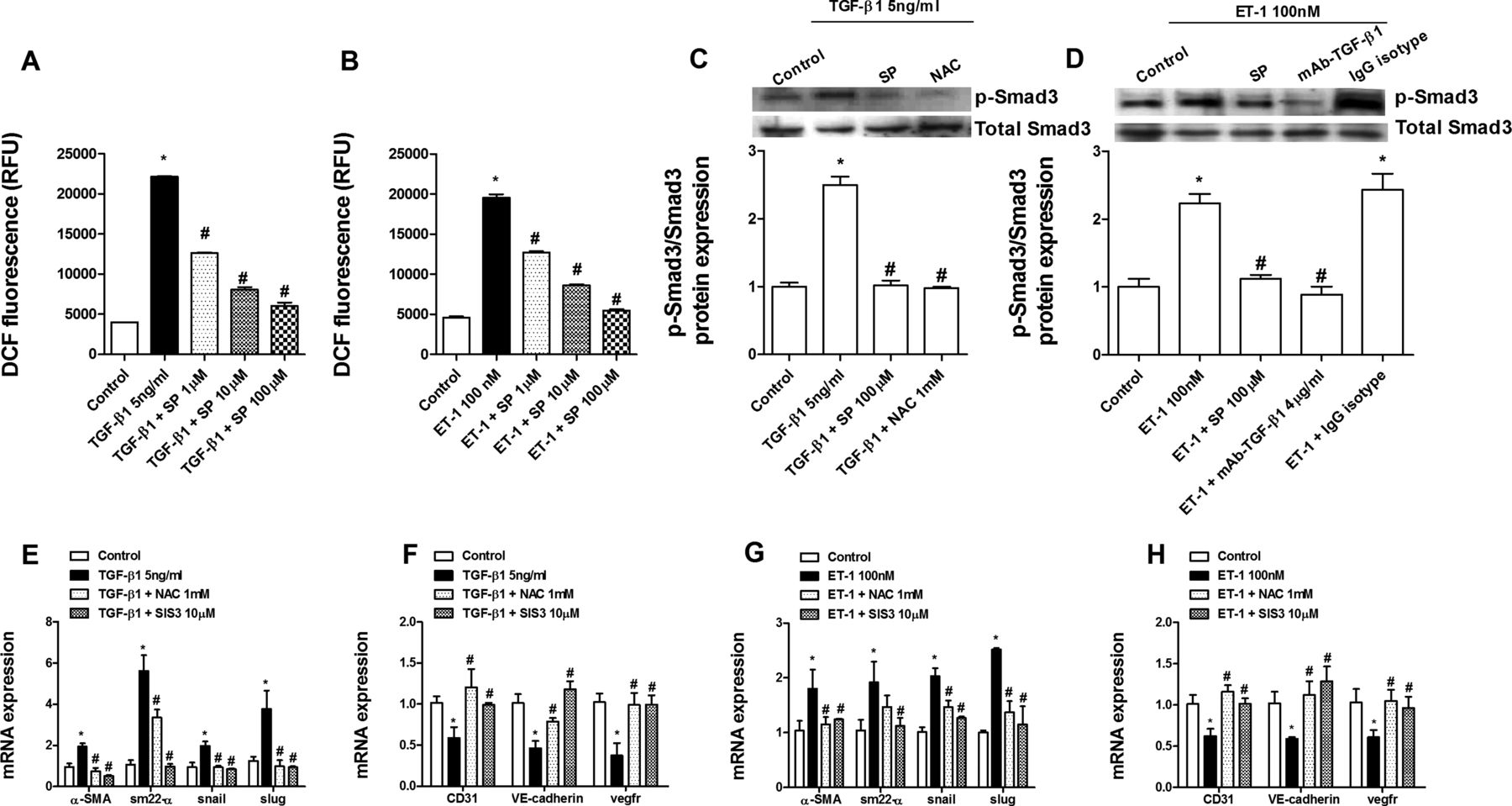
Human pulmonary artery endothelial cells isolated from pulmonary arteries of normal lungs were stimulated with TGF-β1 (5 ng/mL) or ET-1 (100 nM) for 72 h. Both TGF-β and ET-1 elicited an increase in levels of the mesenchymal markers α-smooth muscle actin, SM22-α and its transcriptional regulators snail and slug after 72 h. Similarly, α-smooth muscle actin protein immunofluorescence was also upregulated. In contrast, the endothelial markers CD31, VE-cadherin and vascular endothelial growth factor receptor were downregulated, supporting a change in phenotype (figure 5A–F). Sepiapterin pretreatment dose-dependently inhibited the upregulation of mesenchymal markers and downregulation of endothelial markers induced by both TGF-β1 and ET-1 (figure 5A–F). In other experiments, sepiapterin dose-dependently inhibited intracellular reactive oxygen species formation following TGF-β1 or ET-1 stimulation (figure 6A,B). Furthermore, sepiapterin suppressed TGF-β1- and ET-1-induced Smad3 phosphorylation. The antioxidant N-acetyl-ι-cysteine also suppressed Smad3 phosphorylation (figure 6C). ET-1-induced Smad3 phosphorylation was inhibited by blocking active TGF-β1 in the supernatant with the monoclonal antibody (mAb)-TGF-β1 (figure 6D). As observed for sepiapterin, N-acetyl-ι-cysteine and SIS3, an inhibitor of Smad3, inhibited the increase in levels of mesenchymal markers and decrease in endothelial markers induced by TGF-β1 and ET-1 (figure 6E–H). TGF-β1 and ET-1 decreased eNOS expression and NOx in cell supernatants (figure 7A–C), which were inhibited by sepiapterin and N-acetyl-ι-cysteine. iNOS expression was upregulated in endothelial cells following TGF-β1 and ET-1 stimulation and inhibited by sepiapterin and N-acetyl-ι-cysteine. TGF-β1 and ET-1 also suppressed GTP cyclohydrolase 1 expression but did not affect sepiapterin reductase (figure 7A,B), and sepiapterin and N-acetyl-ι-cysteine prevented GTP cyclohydrolase 1 downregulation. The addition of mAb-TGF-β1 rescued ET-1-induced eNOS and GTP cyclohydrolase 1 downregulation and normalised iNOS expression. Moreover, extracellular nitrotyrosine levels were increased in response to TGF-β1 and ET-1 while culture supernatant levels of BH4, as well as BH4/BH2, were downregulated (figure 7D–F). Sepiapterin administration suppressed nitrotyrosine levels and increased BH4 and BH4/BH2 to near control values (figure 7D–F).

Sepiapterin inhibits endothelial-to-mesenchymal transition in human pulmonary artery endothelial cells (HPAECs). HPAECs were isolated from normal lungs. Cells were incubated with sepiapterin (SP; 1–100 µM) for 30 min before stimulation with (A–C) transforming growth factor β1 (TGF-β1; 5 ng/mL) or (D–F) endothelin 1 (ET-1; 100 nM) for 72 h. Total RNA was isolated for real-time PCR analysis. TGF-β1 and ET-1 upregulated mRNA expression of mesenchymal markers α-smooth muscle actin (α-SMA), SM22-α, slug and snail and downregulated mRNA expression of the vascular endothelial markers CD31, vascular endothelial (VE)-cadherin and vascular endothelial growth factor receptor (VEGFR). (D, E) The addition of mAb-TGF-β1 effectively inhibited the ET-1-induced increase in mesenchymal marker expression and the decrease in vascular endothelial marker expression. (C, F) Phase contrast images and immunofluorescence staining of α-SMA-FITC and DAPI (blue indicates nuclei). Representative images are shown. Scale bar=5 µm. Data are expressed as ratios to GAPDH mRNA normalised to the solvent control group. Results are expressed as means±SE of 3–5 (four cell control population) experiments per condition. Two-way analysis of variance followed by post hoc Bonferroni tests. *p<0.05 vs solvent controls; #p<0.05 vs stimulus.

Sepiapterin inhibition of endothelial-to-mesenchymal transition is mediated by a reduction in reactive oxygen species levels and phosphorylation of Smad3. Human pulmonary artery endothelial cells were incubated with sepiapterin (SP; 1–100 µM), the antioxidant N-acetyl-ι-cysteine (NAC; 1 mM) or the inhibitor of Samd3 (SIS3; 10 µM) for 30 min before stimulation with transforming growth factor β1 (TGF-β1; 5 ng/mL) or endothelin 1 (ET-1; 100 nM) stimulation for (A, B) 30 min, (C, D) 60 min, or (E–H) 72 h. (A, B) Reactive oxygen species were determined by means of DCF fluorescence intensity (in relative fluorescence units (RFU) after TGF-β1 or ET-1 stimulation in the presence or absence of SP. (C, D) TGF-β1 or ET-1 increased the phosphorylation of Smad3 that was inhibited by SP, NAC and mAb-TGF-β1. Phospho-Smad3 and protein levels were expressed as ratios to total Smad3 and normalised to the control group. Representative western blot images are shown (n=4). (E–H) NAC and SIS3 inhibited (E, F) TGF-β1-induced and (G, H) ET-1-induced upregulation of mesenchymal markers and downregulation of endothelial markers. Data in (E–H) are expressed as ratios to GAPDH mRNA levels and normalised to the solvent control group. Results are expressed as means±SE of n=3–5 (four-cell control population) experiments per condition. Two-way analysis of variance followed by post hoc Bonferroni tests. *p<0.05 vs solvent controls; #p<0.05 vs stimulus.
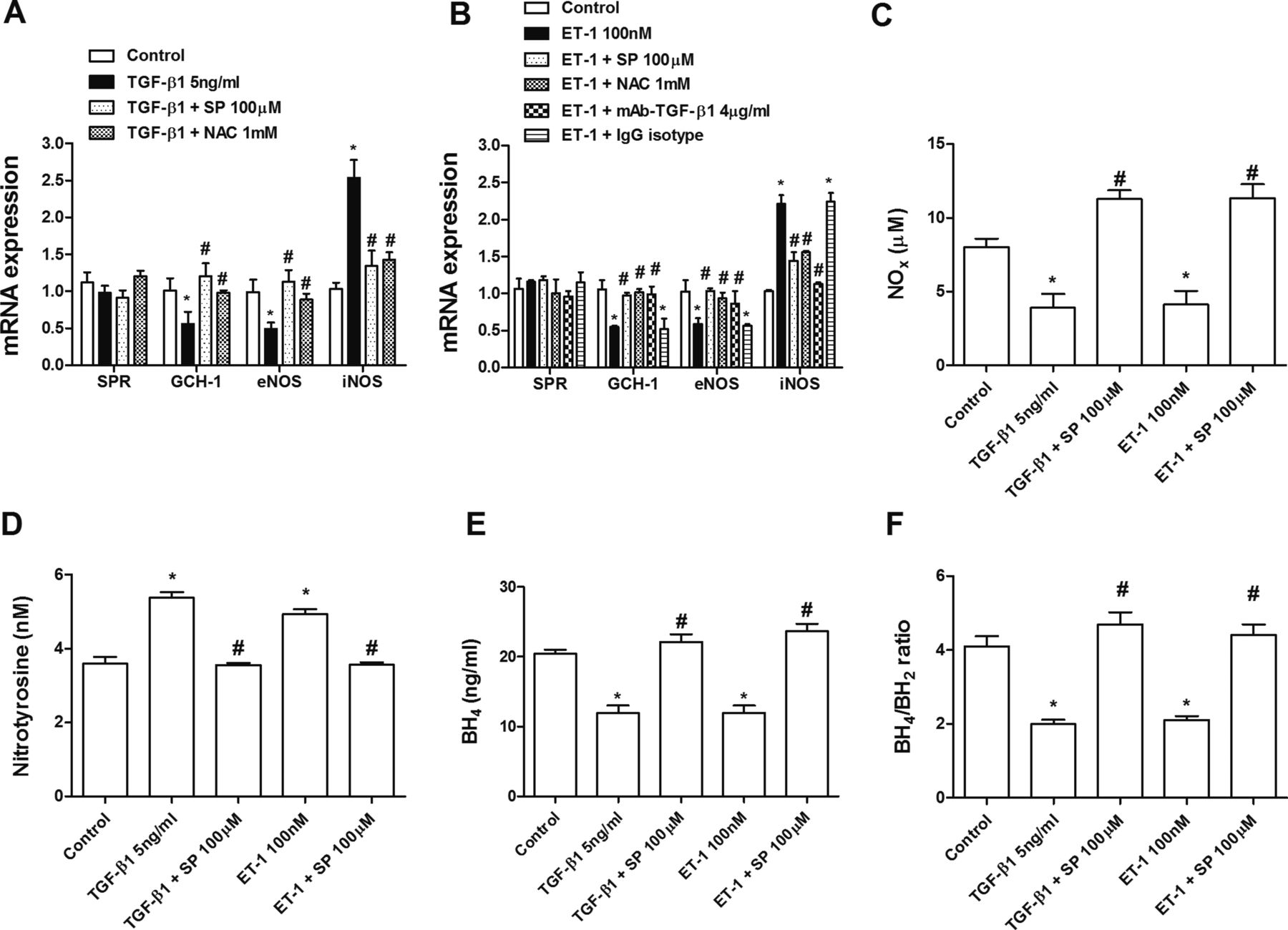
Sepiapterin (SP) improves tetrahydrobiopterin (BH4) bioavailability during endothelial-to-mesenchymal transition. Human pulmonary artery endothelial cells (HPAECs) were incubated with SP (100 µM), N-acetyl-ι-cysteine (NAC; 1 mM), mAb-TGF-β1 (4 µg/mL) or with the IgG isotype control for 30 min and stimulated with (A) transforming growth factor β1 (TGF-β1) or (B) endothelin 1 (ET-1) for 72 h. (A, B) Expression of the sepiapterin reductase (SPR), GTP cyclohydrolase 1 (GCH-1), endothelial nitric oxide synthase (eNOS) and inducible nitric oxide synthase (iNOS) genes in HPAECs. Data are expressed as ratios to GAPDH mRNA levels. Culture supernatant levels of (C) nitrites+nitrates (NOx), (D) nitrotyrosine, (E) BH4 and (F) BH4/BH2 were determined after 72 h of stimulation with TGF-β1 or ET-1 in the presence or absence of SP. Results are expressed as means±SE of n=3–4 (four-cell control population) experiments per condition. Two-way analysis of variance followed by post hoc Bonferroni tests. *p<0.05 vs solvent controls; #p<0.05 vs stimulus.
Sepiapterin inhibits the endothelial-to-mesenchymal transition in vivo
Pulmonary artery sections immunostained with α-smooth muscle actin and VE-cadherin showed an endothelial layer marked with VE-cadherin in control rats and with α-smooth muscle actin and VE-cadherin in bleomycin-treated rats. Oral administration of sepiapterin to bleomycin-treated rats showed an endothelial layer marked by VE-cadherin, suggesting inhibition of endothelial-to-mesenchymal transition in vivo (figure 8). In contrast to normal human pulmonary arteries, those from patients with IPF showed immunostaining with CD31 and collagen type I or with α-smooth muscle actin and VE-cadherin in endothelial cells, as well as in the intima, suggesting a role for endothelial-to-mesenchymal transition in pulmonary remodelling.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Endothelial-to-mesenchymal transition occurs in fibrotic lungs in vivo. Photomicrographs of representative histological sections from (A) pulmonary rat tissue of the control, bleomycin (BL; 3.75 U/kg intratracheally at day 1) and bleomycin plus sepiapterin (BL + SP; 10 mg/kg twice daily orally) groups or (B) pulmonary human tissue from normal lungs (n=6) or idiopathic pulmonary fibrosis (IPF) lungs (n=10). Tissue sections were immunostained with CD31, vascular endothelial (VE)-cadherin, collagen type I or α-smooth muscle actin (α-SMA) antibodies followed by anti-mouse rhodamine or anti-rabbit-FITC secondary antibodies and 4’,6-diamidino-2-phenylindole (DAPI) to stain nuclei. Representative images are shown. White arrows indicate pulmonary artery endothelial cells. IgG isotype controls were negative. Scale bar=30 µm (panel A) or 150 µm (panel B).
Discussion
In this study we provide new evidence of the role of the BH4 system in pulmonary hypertension-associated IPF. BH4, the cofactor of NOS, was decreased in plasma from patients with IPF, contributing to uncoupled NOS activity and an increase in oxidative stress and nitrotyrosine expression. Because pulmonary artery sepiapterin reductase levels were not affected in IPF, oral administration of sepiapterin increased levels of BH4 coupling NOS to produce NO but not peroxynitrite, thereby decreasing pulmonary hypertension and lung fibrosis in bleomycin-treated rats. Furthermore, endothelial-to-mesenchymal transition as a source of fibroblasts was inhibited by sepiapterin in vitro and in vivo. These results provide support for a new therapeutic strategy to attenuate pulmonary hypertension in IPF through the administration of sepiapterin.
The study of NO in IPF lungs is not novel. More than a decade ago Saleh et al10 reported that the lungs of patients with IPF express high levels of iNOS and nitrotyrosine, a product of peroxynitrite, and that eNOS expression was almost absent in the endothelium of pulmonary arteries of patients with IPF, thus contributing to the increased oxidative stress found in IPF. Comparable results were observed in this study. However, oxidative stress in patients with IPF is reportedly not restricted to lung tissue and is also found in serum.17 We identified high levels of nitrotyrosine in the plasma of patients with IPF, which suggests uncoupled NOS activity. In contrast to eNOS, expression of iNOS leads to a 1000-fold increase in levels of NO which mediates defence and pathological processes.18 When iNOS is uncoupled, NO formation is decreased in favour of O2− with which NO reacts rapidly to form peroxynitrite, thus contributing to oxidation. We observed a reduction in plasma BH4 in patients with IPF, which confirmed uncoupled NOS activity. Furthermore, the levels of the BH4 oxidation product BH2 were upregulated. Because BH2 lacks NOS cofactor activity and competes with BH4 for binding to NOS, a decreased ratio of BH4/BH2 is an acceptable measure of uncoupled NOS.19 Low levels of BH4 may result from oxidative degradation to BH2 or by defects in enzymatic de novo synthesis. A combination of the two processes was observed in the patients with IPF recruited in this study as the expression of the de novo synthesis enzyme GTP cyclohydrolase 1 was downregulated in pulmonary arteries without changes in sepiapterin reductase.
Unlike the many studies of NOS, there is little evidence of the role of GTP cyclohydrolase 1 and sepiapterin reductase in pulmonary hypertension and no evidence of pulmonary hypertension secondary to IPF. Mice deficient in GTP cyclohydrolase 1/BH4 have pulmonary hypertension without systemic hypertension,20 and a spontaneously hypertensive rat model exhibits decreased GTP cyclohydrolase 1/BH4 and increased sepiapterin reductase expression in the aorta.21 In contrast, the administration of sepiapterin restored pulmonary artery endothelial function of fetal lambs with persistent pulmonary hypertension.22 In a similar way, a sepiapterin-based strategy improves post-myocardial infarction fibrosis and left ventricular remodelling by activating the BH4 synthesis ‘salvage pathway’ and increasing bioavailable NO predominantly derived from iNOS.12
Sepiapterin may be a more preferable pharmacological tool than BH4 to investigate the role of NOS uncoupling on pulmonary hypertension and IPF progression because it is a stable precursor of BH4 and is more membrane-permeable than BH4.23 We have shown here that sepiapterin reductase expression is not altered in pulmonary arteries from patients with IPF, suggesting that the BH4 synthesis ‘salvage pathway’ remains activated and susceptible to stimulation by exogenous administration of sepiapterin. Recent studies have shown the importance of sepiapterin reductase expression for sepiapterin-inducing BH4 levels.24 Based on these observations, we assessed the beneficial effects of oral sepiapterin in a bleomycin-induced pulmonary fibrosis model, an appropriate model of pulmonary hypertension associated with pulmonary fibrosis.25 As observed in humans with IPF, comparable sepiapterin reductase expression was observed in control and bleomycin-instilled rats while GTP cyclohydrolase 1 and eNOS were downregulated in pulmonary arteries. In contrast, iNOS expression was upregulated, as measured by plasma nitrotyrosine and NOx, suggesting uncoupled NOS. This was confirmed by the reduction in BH4 plasma levels. Supporting the hypothesis that the ‘salvage pathway’ remains activated in pulmonary fibrosis, oral administration of sepiapterin improved pulmonary artery remodelling and hypertension, RV hypertrophy, lung fibrosis extension and rat survival. These events occurred concurrently with coupled NOS function through an increase in BH4/BH2 and a reduction of plasma nitrotyrosine levels, which support similar findings in a mouse model of myocardial infarction.12 However, despite the frequent use of the bleomycin model of pulmonary fibrosis, it has significant limitations in its ability to mimic human IPF since many treatments successful in the bleomycin model were not transferable to human IPF.
To explore possible mechanisms associated with sepiapterin treatment, we studied key molecules related to both pulmonary hypertension and pulmonary fibrosis such as TGF-β1 and ET-1. TGF-β1 and ET-1 share pathological activities in pulmonary hypertension and IPF, such as increasing the profibrotic markers collagen type I or CTGF or inducing fibroblast/myofibroblast transformation. Thus, both TGF-β1 and ET-1 are implicated in the cellular processes of epithelial-to-mesenchymal transition,26 ,27 endothelial-to-mesenchymal transition,28 ,29 fibroblast and smooth muscle proliferation and myofibroblast transformation,5 and in vascular and lung tissue remodelling of patients with IPF. Notably, oral sepiapterin administration inhibited TGF-β1 and ET-1 expression, suggesting an important role in cellular fibroblast-like transformation. Particularly important is the role of NO in cellular transformation. Inhaled NO attenuated extracellular matrix accumulation and the number of lung myofibroblasts and improved endothelial function in patients with IPF with pulmonary hypertension.3 In addition, TGF-β1-induced alveolar epithelial-to-mesenchymal transition was mediated by downregulation of eNOS/NO and inhibited by increasing NO. In a similar way, chronic eNOS inhibition induces endothelial-to-mesenchymal transition in kidney endothelial cells.30 Recent data demonstrated that, in vivo, pulmonary capillary endothelial cells, through endothelial-to-mesenchymal transition, might serve as a source of fibroblasts in pulmonary fibrosis.4 The activity of sepiapterin as an endothelial-to-mesenchymal transition inhibitor may therefore explain its inhibitory effects on pulmonary artery remodelling and lung fibrosis. In this study, TGF-β1 and ET-1 induced endothelial-to-mesenchymal transition in human pulmonary artery endothelial cells. Endothelial-to-mesenchymal transition induced by TGF-β1 was mediated by increased Smad3 phosphorylation and intracellular reactive oxygen species generation, which confirms previous observations.31 ,32 ET-1-induced endothelial-to-mesenchymal transition was provoked by the release of TGF-β1 and its autocrine action, similar to the mechanism underlying ET-1-mediated alveolar epithelial-to-mesenchymal transition.26 As reported previously, TGF-β1 and ET-1 decreased the expression of eNOS30 ,33 but increased the expression of iNOS, which is consistent with a change in phenotype.34 However, nitrotyrosine levels were upregulated in human pulmonary artery endothelial cells, suggesting uncoupled NOS during the endothelial-to-mesenchymal transition process. These results were corroborated by those showing that GTP cyclohydrolase 1 expression and BH4 levels were also reduced after TGF-β1 and ET-1 stimulation. As observed in human pulmonary artery endothelial cells from patients with IPF and in lung endothelial cells from bleomycin-induced pulmonary fibrosis, the expression of sepiapterin reductase was not modified. Thus, administration of sepiapterin to human pulmonary artery endothelial cells cultures inhibited TGF-β1 and ET-1-induced endothelial-to-mesenchymal transition by coupling NOS, decreasing reactive oxygen species and inhibiting phosphorylation of Smad3.
In summary, our findings provide new evidence of the participation of the BH4 system in pulmonary hypertension associated with IPF. Targeting the BH4 synthesis ‘salvage pathway’ by sepiapterin administration may be a new therapeutic strategy to attenuate pulmonary hypertension in IPF and inhibit IPF progression.
Acknowledgments
We thank G Juan and E Guijarro of the Respiratory and Surgery units of the Hospital General, Valencia, Spain for facilitating clinical data and tissue collection.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
PA and JM contributed equally.
-
Contributors PA, JM, JC: conception and design. PA, JM, AD, AS-M, AX, FP-V, AC: acquisition of data. PA, JM, JC: analysis and interpretation of data. JM, AD, AS-M, AX, FP-V, AC, JC: drafting the article or revising it critically for important intellectual content. All authors approved this version of the manuscript.
-
Funding This work was supported by the Spanish government by grants SAF2011-26443 (to JC), FIS CP11/00293 (to JM), CIBERES (CB06/06/0027), ADE10/00020 (to JC), FIS09/00672 (to AX), SAF2010-22066-C02-02 (to AC), research grant from the Valencia Pneumology Foundation 2010 (to AD) and support from the CENIT programme, and research grants from Regional Government (Prometeo/2008/045, ‘Generalitat Valenciana’).
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval This study was approved by the ethics committee of the University General Hospital of Valencia, Spain (CEIC28/2008).
-
Provenance and peer review Not commissioned; externally peer-reviewed.