Article Text

Abstract
Background Pulmonary hypertension (PH) is a common disorder in patients with idiopathic pulmonary fibrosis (IPF) and portends a poor prognosis. Recent studies using vasodilators approved for PH have failed in improving IPF mainly due to ventilation (V)/perfusion (Q) mismatching and oxygen desaturation. Janus kinase type 2 (JAK2) is a non-receptor tyrosine kinase activated by a broad spectrum of profibrotic and vasoactive mediators, but its role in PH associated to PH is unknown.
Objective The study of JAK2 as potential target to treat PH in IPF.
Methods and results JAK2 expression was increased in pulmonary arteries (PAs) from IPF (n=10; 1.93-fold; P=0.0011) and IPF+PH (n=9; 2.65-fold; P<0.0001) compared with PA from control subjects (n=10). PA remodelling was evaluated in human pulmonary artery endothelial cells (HPAECs) and human pulmonary artery smooth muscle cells (HPASMCs) from patients with IPF in vitro treated with the JAK2 inhibitor JSI-124 or siRNA-JAK2 and stimulated with transforming growth factor beta. Both JSI-124 and siRNA-JAK2 inhibited the HPAEC to mesenchymal transition and the HPASMCs to myofibroblast transition and proliferation. JAK2 inhibition induced small PA relaxation in precision-cut lung slice experiments. PA relaxation was dependent of the large conductance calcium-activated potassium channel (BKCa). JAK2 inhibition activated BKCa channels and reduced intracellular Ca2+. JSI-124 1 mg/kg/day, reduced bleomycin-induced lung fibrosis, PA remodelling, right ventricular hypertrophy, PA hypertension and V/Q mismatching in rats. The animal studies followed the ARRIVE guidelines.
Conclusions JAK2 participates in PA remodelling and tension and may be an attractive target to treat IPF associated to PH.
- JAK2
- idiopathic pulmonary fibrosis
- pulmonary hypertension
- BKCa
- Pulmonary artery smooth muscle cells, Pulmonary artery endothelial cells.
Statistics from Altmetric.com
- JAK2
- idiopathic pulmonary fibrosis
- pulmonary hypertension
- BKCa
- Pulmonary artery smooth muscle cells, Pulmonary artery endothelial cells.
Key messages
What is the key question?
To what extent does the Janus kinase type 2 (JAK2) pathway contribute to pulmonary hypertension (PH) associated to idiopathic pulmonary fibrosis (IPF)?
What is the bottom line?
JAK2 is activated in pulmonary arteries from IPF and IPF+PH patients but not in healthy controls, and its inhibition ameliorates pulmonary artery remodelling, increases pulmonary artery relaxation and improves ventilation/perfusion mismatching.
Why read on?
This is the first report that relates JAK2 with IPF and pulmonary artery remodelling identifying a novel target for therapeutics in IPF associated to PH.
Background
Idiopathic pulmonary fibrosis (IPF) is the most common idiopathic interstitial pulmonary disease with a median survival of 2.5–5 years after diagnosis. Pulmonary hypertension (PH), defined by an elevation of mean pulmonary arterial pressure ≥25 mm Hg, in IPF portends a poor prognosis and accelerates lung function decline and death.1 Recent epidemiology studies recognise the prevalence of PH in IPF as 32%–84%, depending on the stage of IPF progression.1
The increase of vascular muscularisation followed by fibrous vascular atrophy and pronounced intimal fibrosis of small pulmonary arteries are the hallmarks of PH in patients with IPF.2 Pulmonary artery remodelling in patients with IPF associated to PH are characterised by different cellular alterations, including endothelial dysfunction,3 the endothelial-to-mesenchymal transition (EnMT) as a source of myofibroblasts,4 as well as pulmonary artery smooth muscle proliferation and transition to myofibroblasts.5
Although PH is clearly associated with worse IPF outcomes, recent clinical trials have showed discouraging results when drugs approved for PH were used to treat IPF. This is the case of ET-1 receptor antagonists, prostacyclin analogues or soluble guanylate cyclase activators that failed in IPF clinical trials.6 Deleterious effects of vasodilators in hypoxic PH, such as IPF, are attributed to increased oxygen desaturation mediated by the propensity to worsen ventilation/perfusion (V/Q) matching by dilating vessels perfusing poorly ventilated lung units.7 Therefore, an ideal strategy to treat IPF associated to PH would be to inhibit lung fibrotic propagation and increase vasodilation preferably in those areas well ventilated but not in poorly ventilated areas.
JAK2/STAT3 pathway is activated in a broad range of fibrotic disorders such as myelofibrosis, skin, liver, myocardial and kidney fibrosis fibrosis.8–12 Janus kinase (JAK)-2 is a key regulator of cytokine signalling, and alterations of JAK2 signalling cause profound changes in response to cytokine stimulation. Transforming growth factor β1 (TGFβ1) signalling can induce phosphorylation and activation of JAK2, which then interacts and phosphorylates STAT3 to induce fibrotic responses.12 Interestingly, JAK2 can be activated by other profibrotic mediators such as plateled-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), interleukin (IL)-6, IL-13, angiotensin II (AT2), 5-HT and ET-1, all of them activated in IPF and PH and with the capacity to produce pulmonary vasoconstriction (the case of 5-HT, ET-1 and AT2).13 14 However, the role of JAK2 in IPF and PH is poorly understood. In contrast, STAT3 phosphorylation has been observed in fibrotic lung tissue from patients with IPF and participates in fibroblast to myofibroblast transition and lung epithelial cell damage being an attractive target to treat IPF.15–17 In this work, we analysed the participation of JAK2 as lung profibrotic marker, activator of pulmonary artery remodelling of IPF patients as well as the potential of JAK2 inhibition as target to treat IPF and the associated PH.
As several JAK2 inhibitors are currently evaluated in clinical trials for malignancies and inflammatory diseases, the antifibrotic effects in experimental models of lung fibrosis associated to PH may have direct translational implications.
Materials and methods
See the online supplementary for a more detailed version of these methods.
Supplemental material
Patients and cell culture
Human lung tissue was obtained from patients with IPF associated to PH (n=9) and IPF (n=10) who underwent surgery for organ transplantation programme, and lung explant healthy control samples were obtained from organ transplant programme from University General Consortium Hospital of Valencia (n=10). Informed written consent was obtained from each participant. See online supplementary for details. Cellular experiments were performed in primary human pulmonary artery endothelial cells (HPAECs) and human pulmonary artery smooth muscle cells (HPASMCs) as previously outlined.18 See online supplementary for further details.
Immunochemistry, immunofluorescence and western blotting
Collagen type I (Col I), α-smooth muscle actin (α-SMA), CD31, JAK2/phospho-JAK2, STAT3/phosho-STAT3, SMAD3/phosphor-SMAD3 and MLCK/phosphor-MLCK were detected in human lung tissue or in pulmonary artery rings by immunohistochemistry, immunofluorescence or western blot as outlined previously.19 Lung slices were stained with Masson’s trichrome to evaluate severity of lung fibrosis that was scored on a scale from 0 (normal lung) to 8 (total fibrotic obliteration of fields) according to Ashcroft score.20 Details are described in the online supplementary.
Real-time RT-PCR and siRNA experiments
Total RNA was obtained from pulmonary arteries or culture cells. The relative quantification of different transcripts was determined using the 2−ΔΔCt method with β-actin as the endogenous control and normalised to a control group, as described previously.21
HPAECs and HPASMCs cells were transfected with siRNA (50 nM) of scrambled siRNA control or with JAK2 gene-targeted siRNA. The transfection reagent used was lipofectamine-2000 (Invitrogen, Paisley, UK) at a final concentration of 2 µg/mL. Details are described in the online supplementary.
Pulmonary artery functional studies, intracellular free Ca2+ measurements and electrophysiological studies
Human precision-cut lung slices from control subjects, IPF and IPF+PH patients were obtained. Slices with pulmonary arteries of approximately 100–300 µm of internal diameter were obtained as previously described.22 Intracellular free calcium concentration ([Ca2+]i) was measured in HPASMCs by epifluorescence microscopy as we previously outlined.23 Membrane currents were recorded in rat PASMCs using the whole-cell configuration of the patch clamp technique as previously outlined.24 Details are described in the online supplementary.
Intratracheal bleomycin animal model
Animal experimentation and handling were performed in accordance with the guidelines of the Committee of Animal Ethics and Well-Being of the University of Valencia (Valencia, Spain). The animal studies followed the ARRIVE guidelines.25 A single dose of 3.75 U/kg bleomycin was administered intratracheally at day 1 of the experimental procedure.19 JSI-124 1 mg/kg/day intraperitoneal (i.p.)26 was administered from day 7 to day 28 of procedure as therapeutic protocol. Pulmonary perfusion (Q), ventilation (V) and the V/Q ratio were analysed using small animal CT (micro-CT) and single-photon emission CT (SPECT; Oncovision micro-CT-SPECT-PET Imaging System; Albira, Valencia, Spain) imaging with the radioisotopes diethylene-triamine-pentaacetate (10 mCi; DTPA-Tc99m) for ventilation and 0.5–1 mCi macroaggregated albumin (MAA)-Tc99m for perfusion, as outlined previously with modifications.18 Details are described in the online supplementary.
Statistical analysis
The Kruskal-Wallis test followed by Dunn’s post hoc tests were used when more than two groups were compared in human studies. Two-tailed Student’s paired t-tests were used to compare two groups of dependent samples in the animal and cell studies and unpaired t-tests for independent samples. Multiple comparisons were analysed by a one-way or two-way analysis of variance followed by Bonferroni post hoc tests. Details are described in the online supplementary.
Results
JAK2 is increased and activated in lungs and pulmonary arteries from patients with IPF
The JAK2 and STAT3 mRNA transcript levels were increased in isolated pulmonary arteries of IPF patients with (2.65 fold; P<0.0001) and without PH (1.94 fold; P=0.0001) compared with control subjects (see clinical data in table 1 and figure 1A). Pulmonary arteries from patients with IPF showed higher JAK2 (39% higher; P<0.0001) and STAT3 (31% higher; P<0.0001) protein expression than in control subjects. The active phosphorylated forms of JAK2 (42% higher; P<0.0001 vs control) and STAT3 (66% higher; P<0.0001 vs control) were upregulated in patients with IPF and significantly overexpressed in pulmonary arteries from IPF+PH patients in the case of JAK2 (70% higher; P=0.0112 vs IPF; figure 1B). Lung fibrotic remodelling markers such as Col I, α-SMA and TGFβ1, and vasoactive profibrotic markers such as ET-1, AT2R and 5-HTR2A were also increased in IPF and IPF+PH pulmonary arteries (figure 1A).
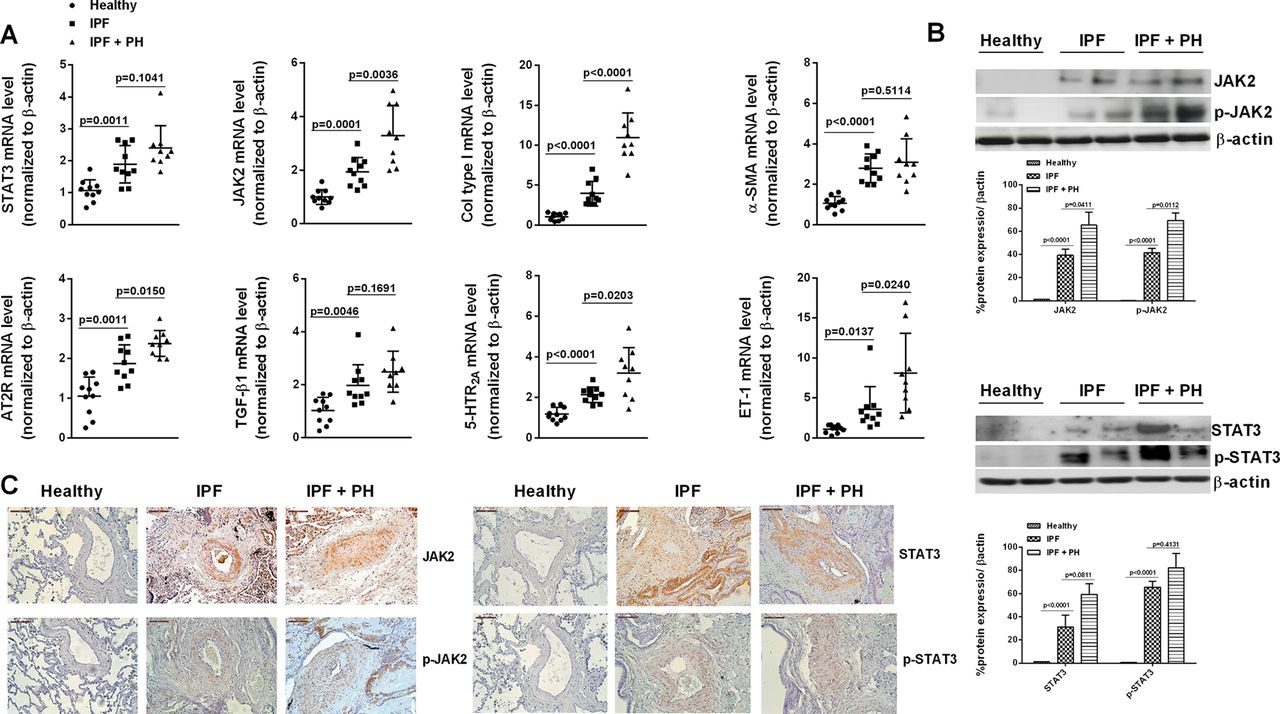
Expression and localisation of JAK2 in pulmonary arteries from patients with idiopathic pulmonary fibrosis (IPF). Isolated pulmonary arteries from control subjects (n=10), IPF (n=10) and IPF+PH patients (n=9) was obtained. (A) Gene mRNA expression of JAK2, STAT3, collagen type I, α-SMA, AT2R, TGFβ1, 5-HTR2A and ET-1 gene expression was analysed by real-time PCR. (B) Protein expression of JAK2/p-JAK2 and STAT3/p-STAT3 analysed by western blot. (C) Immunohistochemistry of JAK2/p-JAK2 and STAT3/p-STAT3. Scale bar: 100 µm. The IgG isotype control was negative (data not shown). Data are expressed as the ratio to β-actin. Data are presented as median with IQR. ‘P’ exact values were obtained following Kruskal-Wallis test. α-SMA, α-smooth muscle actin; JAK2, Janus kinase type 2; PH, pulmonary hypertension; TGFβ1, transforming growth factor β1.
Clinical characteristics
Lung section immunohistochemistry showed a weak JAK2 and STAT3 expression in control subjects, mainly localised in alveolar macrophages (figure 1C), but not detected in pulmonary arteries. In contrast, lung sections from IPF and IPF+PH patients showed elevated expression of JAK2 and STAT3 localised in the intima and media but not in the adventitia of small pulmonary arteries (figure 1C). Phosphorylated forms of JAK2 and STAT3 were located in the cell nucleus of pulmonary artery cells. JAK2, STAT3 and their phosphorylated forms were also detected in fibroblasts and alveolar type II cells from fibrotic areas (online supplementary figure 1).
Supplemental material
JAK2 contributes to pulmonary artery remodelling in ex vivo human pulmonary arteries from patients with IPF
In other ex vivo experiments (see online supplementary for experimental design details), pulmonary artery ring explants from patients with IPF were cultured and stimulated with 5 ng/mL TGFβ1 for 72 hours in the presence or absence of the JAK2 inhibitor JSI-124 (10 nM–1 µM). TGFβ1 treatment resulted in decreased expression of the endothelial markers VE-cadherin (−0.47-fold vs control; P<0.05) and eNOS (−0.46 fold vs control; P<0.05) and the increase of remodelling mesenchymal markers Col I (sixfold vs control; P<0.05) and α-SMA (3.5-fold vs control; P<0.05 (figure 2A) in both endothelial cells and cells located in the intima (figure 2B). JSI-124 inhibited the effect of TGFβ1 on the loss of endothelial markers and upregulation of the pulmonary artery remodelling markers at 100 nM–1 µM (figure 2A,C), suggesting pulmonary artery antiremodelling properties in patients with IPF.

JAK2 inhibition reduced transforming growth factor (TGF)β1-induced ex vivo PA ring remodelling. PA rings from (A and B) patients with IPF (n=4) were incubated with the JAK2 inhibitor JSI-124 (10 nM–1 µM) for 1 hour and then stimulated with 5 ng/mL TGFβ1 for 72 hours. Artery rings were homogenised to extract RNA for gene expression analysis (A), embedded in paraffin for immunofluorescence analysis (B) or western blot analysis (C). Scale bar: 150 µm. Yellow arrows indicate adventitial and smooth muscle layers. White arrows indicate endothelial cells. Four arterial rings per patient were used. The results are shown as mean±SE Two-way analysis of variance followed by Bonferroni post hoc tests. *P<0.05 compared with control; #P<0.05 compared with TGFβ1. α-SMA, α-smooth muscle actin; JAK2, Janus kinase type 2; IPF, idiopathic pulmonary fibrosis; PA, pulmonary artery; TGFβ1; transforming growth factor β1.
JAK2 mediates TGFβ1-induced pulmonary artery endothelial-to-mesenchymal and smooth muscle cell to myofibroblast transitions
The next experiments were designed to explore the effects of JAK2 on remodelling alterations of pulmonary arteries of IPF patients, including HPAECs and HPASMCs to mesenchymal transition. Incubating the HPAECs with TGFβ1 changed their endothelial phenotype to a mesenchymal/myofibroblast phenotype characterised by loss of the endothelial markers VE-cadherin, VEGFR1, FVIII and eNOS and an increase in the mesenchymal markers Col I, α-SMA and vimentin (figure 3A). JAK2 gene silencing (siRNA-JAK2) or pharmacological inhibition of JAK2 using JSI-124, prevented these changes and maintained the endothelial phenotype (P<0.05; figure 3A). TGFβ1 increased the expression of collagen type I and vimentin in HPASMCs; this effect was inhibited by siRNA-JAK2 and JSI-124 treated cells (P<0.05; figure 3B). Furthermore, siRNA-JAK2 and JSI-124 treatments inhibited TGFβ1-induced HPASMC proliferation (P<0.05; figure 3C). Adding 5 ng/mL TGFβ1 to HPAECs and HPASMCs from patients with IPF increased the phosphorylation of JAK2 and SMAD3, which was inhibited by siRNA-JAK2 and JSI-124 (figure 3D,E).

Effects of JAK2 inhibition on TGFβ1-induced endothelial-mesenchymal and smooth muscle cell-mesenchymal transition. (A) Human PA endothelial cells (HPAECs) and (B and C) human PA smooth muscle cells (HPASMCs) were isolated from PAs of patients with IPF (n=4). The cells were incubated with JSI-124 (1 µM) for 30 min or treated with siRNA to silence JAK2 gene. Cells were then stimulated with 5 ng/mL TGFβ1 for (A) 72 hours or (B) 48 hours. Endothelial and mesenchymal marker gene mRNA expression was analysed. (C) HPASMCs from patients with IPF were incubated with JSI-124 (1 µM) for 30 min or treated with siRNA to silence JAK2 gene, and then stimulated with 5 ng/mL TGFβ1 for 48 hours in 96-well plates to measure cell proliferation with the bromodeoxyuridine (BrdU) assay. HPAECs (D) and HPASMCs (E) were incubated with JSI-124 (1 µM) for 30 min or treated with siRNA to silence JAK2 gene, and then stimulated with 5 ng/mL TGFβ1 for 1 hour. Protein expression of total Jak2 and Smad3, and phosphorylated Jak2 and Smad3 were analysed together with the internal control β-actin by western blot. The results are shown as mean±SE of n=4 cell populations from patients with IPF. (F) Immunofluorescence for α-SMA in HPAECs stimulated with 5 ng/mL TGFβ1 in presence or absence of JSI-124 (1 µM). (G) Discs with cultured HPAECs were incubated for 72 hours with 5 ng/mL TGFβ1 in presence or absence of JSI-124 (1 µM). Then, a gel disk with cultured HPAECs was placed in the microscope and cells imaged with bright field illumination. The graph shows the time course of contractile response of HPAECs challenged with ET-1 (100 nM). Values are expressed as total force exerted by the cell on the substrate. Two-way analysis of variance followed by Bonferroni post hoc tests. *P<0.05 compared with control; #P<0.05 compared with TGFβ1. α-SMA, α-smooth muscle actin; JAK2, Janus kinase type 2; PA, pulmonary artery; TGFβ1; transforming growth factor β1.
To analyse the phenotype characteristics of endothelial to mesenchymal transition, endothelial cells were immunostained with α-SMA antibody. TGFβ1 produced α-SMA fibres that were inhibited by JSI-124 (figure 3F). Formation of α-SMA fibres increased the capacity of cell contraction as assessed by traction microscopy (figure 3G) confirming the mesenchymal/myofibroblast phenotype (figure 3G).
Pulmonary arteries from healthy controls showed a lack of JAK2, p-STAT3 and α-SMA expression and positive immunostaining for CD31 in endothelial layer (online supplementary figures 2 and 3). In contrast to normal human pulmonary arteries, those from IPF and IPF+PH patients showed coimmunostaining with CD31 and JAK2/p-STAT3 and with α-SMA and JAK2/p-STAT3 in endothelial cells, as well as in the intima, suggesting a role for EnMT in pulmonary remodelling (online supplementary figures 2 and 3).
JAK2 inhibition promotes relaxant and anticontractile effects on pulmonary arteries from patients with IPF
JSI-124 dose-dependently relaxed 5-HT precontracted pulmonary arteries to nearly 80% of maximum inhibition at 10 µM (figure 4A). JSI-124 induced a direct relaxation on basal tone of IPF pulmonary arteries, near to 40% of maximum relaxant effect of papaverine (figure 4B). Pulmonary artery relaxant effects of JSI-124 were higher in pulmonary arteries from control subjects (EC50 6.5±0.15 µM, maximal inhibition, 78%±6.4%; P<0.05 vs IPF and IPF+PH; figure 4C) than in IPF (EC50 5.9±0.12 µM, maximal inhibition, 70%±2.6%; P<0.05 vs control; figure 4C) and IPF+PH (EC50 5.8±0.1 µM, maximal inhibition, 55%±5.2%; P<0.05 vs control; figure 4C). In order to study the contribution of the endothelium in vascular relaxation induced by JSI-124, pulmonary arteries from patients with IPF were pretreated with L-Noarg 1 µM or with indomethacin. Neither L-Noarg nor indomethacin were able to reduce relaxant effects of JSI-124 (figure 4D). To analyse the contribution of potassium channels to the relaxant effects of JSI-124, cut lung slices were treated with tetraethylammonium (a non-selective potassium channels blocker), iberiotoxin (IBTX; an inhibitor of large conductance calcium-activated potassium channel (BKCa)) or charybdotoxin (an inhibitor of large conductance calcium-activated potassium channels and slowly inactivating voltage-gated potassium channels) before JSI-124. All potassium inhibitors reduced ~55% relaxation effects of JASI-124, with the same potency and maximum effect, suggesting the participation of BKCa channels in the relaxation process. To evaluate the effect of the JAK2 inhibitor on hypoxic pulmonary vasoconstriction (HPV), lung cut slices were incubated in hypoxic conditions (95% N2–5% CO2) during 1 hour, inducing a 25%–35% of the maximal response induced by 80 mM KCl (data not shown). After 1 hour of hypoxic conditions, JSI-124 showed a vasodilator effect that reach 50% in control arteries and 22% in IPF arteries, suggesting a predominant HPV inhibitor role in control arteries rather than in IPF arteries.

Relaxant effects of JAK2 inhibition on pulmonary arteries from patients with idiopathic pulmonary fibrosis (IPF). (A) Concentration-dependent relaxant curves of the JAK2 inhibitor JSI-124 on small pulmonary arteries precontracted with 10 µM serotonin (5-HT) in precision lung cut slices from IPF (n=6). (B) Direct relaxing effect of JSI-124 on basal tone of small pulmonary arteries of precision lung cut slices from IPF (n=6). (C) Concentration-dependent relaxant curves of the JAK2 inhibitor JSI-124 on small pulmonary arteries precontracted with 5-HT 1 µM in precision lung cut slices from control donors (n=10), IPF (n=6) and IPF+PH (n=6). (D) Precontracted 5-HT pulmonary arteries from cut lung slices of patients with IPF (n=6) were incubated with the endothelial nitric synthase inhibitor L-Noarg, the cyclooxygenase inhibitor indomethacin, (E) with tetraethylammonium (TEA; a non-selective potassium channels blocker), iberiotoxin (IBTX; an inhibitor of large conductance calcium-activated potassium cannel (BKCa)) or charybdotoxin (ChTX; an inhibitor of large conductance calcium-activated potassium channels and slowly inactivating voltage-gated potassium channels) for 15 min, followed by the addition of growing concentrations of JSI-124. (F) Lung cut slices were incubated in hypoxic conditions (95% N2–5% CO2) during 1 hour, and cumulatively increasing concentrations of JSI-124 (from 0.01 nM to 50 µM) were added in control (n=6) and IPF (n=6) arteries. (G and H) Pulmonary arteries from precision lung cut slices from control donors (n=10) and patients with IPF (n=6) were incubated with JSI-124 for 30 min followed by increasing 5-HT concentrations. The results are shown as mean±SE. Two-way analysis of variance followed by Bonferroni post hoc tests. *P<0.05 versus control. JAK2, Janus kinase type 2; PH, pulmonary hypertension.
In other experiments, JSI-124 significantly inhibited 5-HT-induced contractions in control donors and patients with IPF (figure 4F,G). Importantly, the inhibitory effect of JSI-124 on 5-HT-induced contraction was significantly higher in control subjects than in IPF cut lung slices (figure 4F,G; P<0.05).
JAK2 inhibits BKCa potassium currents and increases intracellular Ca2+ in pulmonary artery smooth muscle cells
Using electrophysiology records, we observed that JSI-124 1 µM increased BKCa currents in isolated rat PASMCs (P<0.05 vs control; figure 5A,B). The BKCa inhibitor IBTX, suppressed JSI-124-induced K+ currents (figure 5C,D) suggesting that JAK2 modulates the closure of BKCa and that the inhibition of JAK2, using JSI-124, reactivates BKCa and induces vascular relaxation. The inhibition of BKCa channels increased cytoplasmic Ca2+ as we observed using IBTX 100 nM as stimulus in HPASMCs from patients with IPF (figure 5F,G). IBTX-induced cytoplasmic Ca2+ was inhibited in cells preincubated with JSI-124 and in cells treated with siRNA-JAK2 (P<0.05 vs IBTX stimulus; figure 5F,G). The increase of MLCK phosphorylation induced by 5-HT in HPAECs from IPF patients was inhibited by JSI-124 as assessed by western blot and immunofluorescence (figure 5H,I). The expression of the BKCaα1 and BKCaβ1 decreased in pulmonary arteries from IPF and IPF+PH compared with control arteries (figure 5J,K).

JAK2 inhibition augments BKCa currents in pulmonary artery smooth muscle cells (PASMC). (A–E) Electrophysiological studies performed in PASMCs of rat pulmonary arteries (250–350 µm internal diameter) isolated from male Wistar rats. (A) Representative current traces for 200 ms depolarisation pulses from −60 mV to +60 mV in 10 mV increments from a holding potential −60 mV before (control) and after JSI-124 1 µM. (B) Current–voltage relationships of K+ currents measured at the end of the pulse before (control) and after the addition of JSI-124 (n=5). Representative current traces (C) and current–voltage relationships (D) showing the lack of effect of JSI-124 in the presence of iberiotoxin (IBTX, 0.1 µM, n=5). (E) JSI-124-sensitive current obtained by subtracting the current in the presence and in the absence of the drug in cells perfused with or without (control) IBTX. Results are means±SE. * and ** indicate P<0.05 and P<0.01 versus control, respectively (paired or unpaired Student’s t test). (F and G) Intracellular Ca2+ measured in human PASMCs from patients with IPF patients (n=3) using fura-2AM as Ca2+ indicator in a fluorescence microscopy. Cells were incubated with (F) JSI-124 (10 nM-1µM, 30 min before stimulus) or (G) gene JAK2 silencing and stimulated with iberiotoxin (IBTX). Results are the mean±SE of n=20 cells per condition. *P<0.05 versus stimulus. (H and I) HPASMCs from patients with IPF (n=4) incubated with JSI-124 and stimulated with serotonin (5-HT) 1 µM for 30 min. Levels of phosphorylated (H) and non-phosphorylated (I) myosin light-chain kinase were measured by immunofluorescence (H) and western blot (I). (J and K) Gene mRNA expression of BKCaα1 and BKCaβ1 was measured in pulmonary arteries from control subjects (n=10), IPF (n=10) and IPF+PH patients (n=9). Data are presented as median with IQR. ‘P’ exact values were obtained following Kruskal-Wallis test. BKca, calcium-activated potassium channel; JAK2, Janus kinase type 2.
JAK2 mediates lung fibrosis, PA remodelling and hypertension in rats treated with intratracheal bleomycin
Following 28 days of intratracheal bleomycin instillation, a marked lung fibrotic response was observed as it indicated the increase of Ashcroft score of lung histology (P<0.05 vs control; figure 6A,B). JSI-124 alleviated histologically observed multifocal fibrotic lesions, resulting in fewer organised and smaller foci and reduced septal enlargement with a diminished Ashcroft fibrosis score (P<0.05 vs bleomycin group; figure 6A,B).

Bleomycin-induced lung fibrosis and pulmonary artery remodelling and hypertension is attenuated by JAK2 inhibition. Wistar rats received a single intratracheal dose of bleomycin (BLM; 3.75 U/kg) on day 1. JSI-124 (1 mg/kg/day i.p.) or vehicle was administered from day 7 until analysis at day 28 (n=10 per group). (A) Masson’s trichrome (scale bar: 100 µm) of controls, BLM and BLM+JSI-124. (B) Fibrosis Ashcroft scores were assessed as described in the Methods. (C) Ventricular right hypertrophy, (D) pulmonary artery remodelling and (E) pulmonary artery pressure were measured in different groups at the end of the experimental procedure. (F–H) p-Smad, p-Stat and p-Jak2 western blots of lung tissue from control vehicle, BLM and BLM+JSI-124 groups. Data are expressed as the ratio to β-actin for %protein levels. Results are expressed as means±SE, n=10. One-way analysis of variance followed by post hoc Bonferroni tests. *P<0.05 related to controls; #P<0.05 related to BLM. i.p., intraperitoneal; JAK2, Janus kinase type 2; RVSP, right ventricular systolic pressure.
Right ventricular (RV) hypertrophy (RV/left ventricular (LV)+septum) and pulmonary vascular remodelling developed following bleomycin treatment (figure 6A, C and D). By day 28, PH was observed in the bleomycin group, as peak RV systolic pressure had increased from 20±3.6 in control rats to 40.2±4.1 mm Hg in the bleomycin group (P<0.05 vs control; figure 6E). JSI-124 reduced RV hypertrophy, pulmonary vascular remodelling, and normalised PH to 24.5±5.9 mm Hg at day 28 (P<0.05 vs bleomycin group; figure 6E). JSI-124 also reduced collagen deposition, visualised by Masson’s trichrome staining, and muscularisation in the walls of pulmonary arteries (P<0.05 vs bleomycin group; figure 6A,B). Bleomycin induced JAK2, STAT3 and SMAD3 lung phosphorylations at day 28 that were inhibited by JSI-124 (P<0.05 vs bleomycin group; figure 6F–H).
PA sections immunostained with α-SMA, JAK2, p-STAT3 and CD31 showed an endothelial layer marked with CD31 in control rats, and with α-SMA, CD31 and JAK2/p-STAT3 in bleomycin-treated rats (online supplementary figures 4 and 5). Administration of JSI-124 to bleomycin-treated rats showed an endothelial layer marked by CD31, suggesting the inhibition of endothelial-to-mesenchymal transition in vivo (online supplementary figures 4 and 5).
Pulmonary ventilation and perfusion were significantly reduced in the bleomycin-treated group between day 7 and day 28 of the experimental procedure (88% ventilation reduction and 69% of perfusion reduction at day 28 (figure 7A,B). The JAK2 inhibitor JSI-124 was administered between day 7 and day 28 of the experimental procedure and improved pulmonary ventilation and perfusion in a similar degree between day 14 and day 28 (45% of ventilation reduction and 41% of perfusion reduction at day 28; P<0.05 vs bleomycin group; figure 7A,B). The relation between V/Q was analysed at the end of experimental procedure (day 28). V images were taken after breathing DTPA-Tc99m, and Q images were taken after perfusing MAA-Tc99m. As expected, the V/Q ratio was clearly impaired in the lungs of bleomycin-treated rats compared with that in control animals (73.7% of reduction at day 28 vs control; P<0.05 vs control; figure 7C–G). Therapeutic administration of JSI-124 1 mg/kg/day from day 7 to day 28 improved the correlation of V/Q (8.8% of reduction at day 28 vs bleomycin; P<0.05 vs bleomycin group; figure 7C). To compare with other vasodilators approved for PH, bosentan was administered orally at 100 mg/kg between days 7 and 28 of the experimental procedure. Bosentan significantly improved lung perfusion (figure 7E). However, lung ventilation did not show a proportional improvement (figure 7D), so V/Q was impaired. These results were supported by the analysis of arterial blood O2 pressure (PaO2), arterial blood CO2 pressures and the alveolar–arterial O2 pressure difference (AaDO2). JSI-124 improved PaO2 and restored AaDO2 to control values. In contrast, bosentan did not improve PaO2, sustaining elevated AaDO2 values that reflect uncoupled V/Q (figure 7H).

{kind=link}
![[SP2.jpg]](https://thorax.bmj.com/content/thoraxjnl/73/6/519/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
JAK2 inhibition improves pulmonary ventilation, perfusion and ventilation/perfusion (V/Q) mismatch in a rat model of pulmonary fibrosis and pulmonary hypertension induced by bleomycin. A single 3.75 U/kg dose of BLM or vehicle was administered intratracheally on day 0. (A–C) JSI-124 (1 mg/kg/day i.p.) or (D–F) bosentan (100 mg/kg/day oral) were administered from day 7 to day 28. Lung ventilation was measured by DTPA-Tc99m inhalation, and perfusion was measured by injection of MAA-Tc99m via the tail vein at days 7, 14 and 28 of the experimental procedure. Lung signal was measured using micro-CT-coupled to single-photon emission CT. (C andF) The relationship between the ventilation (V) and perfusion (Q) data was determined using PMOD software to analyse the intensity of radiation (arbitrary units) of each volume of interest. (A–F) The figures show the mean±SE of the corrected radiation intensity of the radiation signal of each pixel of the 250 images of V and Q. (G) Representative V and Q images. (H) Arterial blood gas pressures measured at day 28 of the experimental procedure. Results were analysed in 10 animals/experimental condition. One-way analysis of variance followed by Bonferroni post hoc tests. *P<0.05 compared with control; #P<0.05 compared with BLM. AaDO2, alveolar– arterial O2 pressure difference; BLM, bleomycin; i.p., intraperitoneal; JAK2, Janus kinase type 2; PaCO2, arterial blood CO2 pressures; PaO2, arterial blood O2 pressure.
Discussion
In this study, we analysed the effects of JAK2 on pulmonary fibrosis, pulmonary artery remodelling and contraction in an ex vivo/in vitro system of pulmonary arteries from patients with IPF with and without severe PH. This work provide novel evidence of the overexpression of JAK2 and its phosphorylated form in lung tissue and pulmonary arteries from IPF and IPF with severe PH. JAK2-mediated pulmonary artery remodelling in IPF, almost in part, by cell transformations such as HPAEC to mesenchymal transition, and HPASMC to myofibroblast transition and proliferation. JAK2 contributed to 5-HT-induced contraction in small human pulmonary arteries of patients with IPF through the inactivation of BKca channels, the increase of cytoplasmic Ca2+ and the phosphorylation of MLCK in HPASMCs. The animal in vivo model of bleomycin-induced lung fibrosis and PH demonstrated that JAK2 pharmacological inhibition improves lung tissue fibrosis, right ventricular hypertrophy, pulmonary vascular remodelling and hypertension, improving pulmonary perfusion and V/Q mismatching. Our findings may add scientific value to support treatment of IPF and the associated PH using JAK2 inhibitors.
Previous reports have demonstrated the overexpression and activation of STAT3 in lungs from patients with IPF15 16 and patients with PH.27 STAT3 is a cytoplasmic latent transcription factor that is activated by phosphorylation in response to cytokines such as IL-6, growth factors such as PDGF and agonists such as ET-1 and AT2. Src and JAKs are among the proteins the are most frequently involved in the transduction of the signal between the fixation of the agonist on the receptor and the phosphorylation of STAT3, although many others can activate STAT3 such as G-protein coupled receptors agonists.27 Interestingly, p-JAK2 can activate STAT3 and it can activate different intracellular receptors and form multiprotein complexes.28 However, the exact role of p-JAK2 in IPF remains to be dissected. In this work, we observed an increased expression of STAT3, JAK2 and its phosphorylated forms in lung tissue and pulmonary arteries from IPF and IPF+PH patients. While the expression of STAT3 was similar in IPF and IPF+PH groups, the expression of JAK2/p-JAK2 was significantly overexpressed in IPF+PH pulmonary arteries suggesting a dominant role on pulmonary artery remodelling. The p-JAK2 nuclear localisation in pulmonary arteries and fibrotic areas suggest a role as non-canonical transcription factor independent of canonical STAT3 pathway. Previous reports have identified nuclear localisation of p-JAK2 supporting our findings.28 29 Emerging evidence indicates that the nuclear role of p-JAK2 may be of particular significance under physiological and pathological conditions of heightened cellular growth independently of STAT3 activation.28
Some studies have indicated the implication of JAKs proteins in PH, analysing the increase of JAKs mRNA levels in rats with PH induced by hypoxia30 or through the beneficial effect of the JAK2 inhibitor AG490 in reversing PAECs proliferation.31 However, other studies failed to determine JAK2 upregulation/activation in HPASMC of patients with pulmonary artery hypertension (PAH), compared with healthy HPASMCs.32 Overexpression of p-JAK2 has been observed in cytoplasm of skin fibroblasts from patients with systemic sclerosis (SSc)12 and increased especially when associated with PAH, compared with controls and idiopathic PAH where JAK2 levels were not affected.33 However, the state of phosphorylation of JAK2 has not been measured in these studies, poorly allowing any conclusion on whether JAK2 is activated.
However, the increase of JAK2 expression and activation in IPF and to a greater extent in IPF+PH could have different meanings. In this respect, pharmacological inhibition of JAK2 and gene silencing of JAK2 inhibited EnMT and HPASMC to myofibroblast transition and proliferation. EnMT has been proposed as cellular mechanism to increase the number of lung myofibroblasts from endothelial cells to increase lung fibrosis in animal and human studies.4 19 Recent studies indicate that EnMT can be mediated through the activation of JAK/STAT3 pathway in endothelial cells from different types of cancer.34 Moreover, TGFβ1 activates JAK2 and STAT3 in SSc, and pharmacological or genetic inactivation of JAK2 reduces the skin profibrotic effects of TGFβ1.12 In the present work, we showed first evidence of the phosphorylation of JAK2 after TGFβ1 stimulation in both HPAEC and HPASMC of patients with IPF. Furthermore, pharmacological inhibition of JAK2 or gene silencing of JAK2 reduced the TGFβ1-induced SMAD3 phosphorylation that connect TGFβ1 with JAK2 pathway as previously outlined.35 Supporting these results, recent evidence indicates that TGFβ1 can activate STAT3 via SMAD2/3-dependent mechanism in fibroblasts from patients with IPF.16
PA remodelling in PH associated to hypoxaemic lung diseases such as chronic obstructive pulmonary disease or IPF is characterised by remodelling of the intimal and, in a lesser extent, medial layer of muscular arteries but not in adventitia.36 The high expression of JAK2/STAT3 in the intimal layer and media may reflect the active, proliferative and migratory phenotype of intimal resident cells that are mainly myofibroblasts.37 Therefore, the implication of JAK2 in EnMT and smooth muscle cell to myofibroblast transition could link the intimal and media remodelling and the high amount of myofibroblasts in these artery layers.
In addition to TGFβ1, other profibrotic factors such as PDGF, VEGF, IL-6, IL-13, AT2, 5-HT and ET-1 can phosphorylate JAK213 14 and are elevated in pulmonary arteries from IPF and IPF+PH patients as we showed in this work and previously reported.18 In addition to pulmonary fibrosis, 5-HT, ET-1 and AT2 promotes pulmonary artery vasoconstriction, pulmonary remodelling and PH in addition to lung fibrosis.38 39 Previous reports have shown that JAK2 inhibition can reduce intracellular Ca2+ and rat aortic rings contraction induced by 5-HT, ET-1 and AT2.38 39 In this work, we showed first evidence on the role of JAK2 in pulmonary vasoconstriction of small pulmonary arteries from control subjects, IPF and IPF+PH patients. It is known that human pulmonary artery vascular remodelling occurs on small resistant-type intrapulmonary vessels that form part of the pulmonary vascular bed responsible for the pressure elevation observed in PH.40 In this work, the inhibition of JAK2 using JSI-124 as pharmacological approach relaxed 5-HT precontracted small pulmonary arteries of patients with IPF. Furthermore, JSI-124 had direct relaxing effects on untreated basal pulmonary arteries, which suggest a role of JAK2 maintaining basal tone of IPF pulmonary arteries. Interestingly, vascular relaxing effects of JSI-124 were more potent in small pulmonary arteries from control patients than in IPF and IPF+PH, respectively, suggesting a preference of relaxation depending on the nature/remodelling status of pulmonary arteries. Pulmonary artery relaxation induced by JSI-124 was independent of the pulmonary endothelium but dependent of potassium channels. In fact, IBTX, an inhibitor of large conductance calcium-activated potassium channel BKCa, reduced the relaxing effects of JSI-124. Electrophysiological experiments using patch clamp technique showed that JAK2 inhibition increased BKCa currents and that JAK2 inhibition or gene silencing reduced the increase of intracellular Ca2+ induced by BKCa blockage.
A feature of all contractile HPASMCs is the abundant expression of BKCa channels that are voltage dependent, increasing in activity in response to membrane depolarisation.41 A function of the BKCa channels is to provide negative feedback against depolarisation, limiting Ca2+ influx through CaV1.2 (L-type voltage-gated Ca2+) channels. Therefore, the dominant channels in regulation of vascular tone are BKCa channels of the HPASMCs.
In addition, and of direct relevance to microvascular smooth muscle, BKCa is activated by both Ca2+ and voltage to act as a negative feedback control mechanism for contractile stimuli while being sensitive to a number of metabolic stimuli such as partial pressure of oxygen, reactive oxygen species, phosphorylation by protein kinases, steroid hormones and fatty acids and their metabolites. Importantly, a number of vasodilator and vasoconstrictor stimuli use phosphorylation-mediated mechanisms to regulate ion channel activities. In general, PASMC BKCa is activated by cAMP/PKA42 and cGMP/PKG42 while being inhibited by PKC43 and the tyrosine kinase c-Src.44 Also, hypoxic conditions downregulate the expression and activity of BKCa channels that contribute to the elevated pulmonary vascular tone found in PAH.45 In fact, genetic deletion of the β1 subunit of BKCa in mice leads to hypertension and increased contractility of vascular smooth muscle cells.46 47 Furthermore, transformed and dedifferentiated smooth muscle cells into proliferative remodelling phenotype are accompanied by the loss of BKCa channel expression, thus increasing pulmonary wall remodelling.48
In this regard, results obtained in this study showed a decreased expression of BKCaα1 and BKCaβ1 in pulmonary arteries from IPF, and in a greater extent in IPF+PH pulmonary arteries, which may explain, almost in part, the lowest relaxing effects of JSI-124 in pulmonary arteries from IPF and IPF+PH patients compared with control subjects and also the lowest effect of JASI-124 inhibiting hypoxic pulmonary vasconstriction in IPF pulmonary arteries.
These results are of potential value, since pulmonary vasodilation in poorly ventilated areas, as occurs in IPF, increase V/Q mismatch and oxygen desaturation. This is the case of vasodilators approved in PAH and used to treat PH in IPF. In this work, we observed an overexpression and activation of JAK2 in pulmonary arteries from IPF and IPF+PH patients, contributing to pulmonary artery remodelling. Low expression of BKCa in pulmonary arteries from IPF+PH indicated a preferental relaxation in well-ventilated alveolar units. To directly analyse the role of the JAK2 inhibitor JSI-124 on the V/Q matching, we used V/Q micro-CT-SPECT imaging, a well-established nuclear medicine technique that provides spatial information of respiratory gas exchange, ventilation of alveolar units and perfusion of the pulmonary capillary beds.18 Therapeutic administration of JSI-124 from day 7 to day 28 of the experimental procedure improved rat pulmonary artery remodelling, right ventricle hypertrophy, PH, ventilation, lung blood perfusion and V/Q mismatching induced by bleomycin. In contrast, bosentan predominantly increased perfusion and in a lesser extent ventilation, showing a mild impairment of V/Q as previously outlined.49 Although results provided in this work relates JAK2 with IPF and PH, it should be noted that human samples used are from no more than 10 patients, a small sample that can limit the interpretation of this work.
To our knowledge, this is the first report that provides evidence on the JAK2 participation in lung fibrosis and pulmonary remodelling and vasoconstriction of IPF and IPF+PH patients that may be of potential value since JAK2 inhibitors are already in clinical use for other indications.
Supplemental material
![[SP3.jpg]](https://thorax.bmj.com/content/thoraxjnl/73/6/519/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
Supplemental material
![[SP4.jpg]](https://thorax.bmj.com/content/thoraxjnl/73/6/519/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
Supplemental material
![[SP5.jpg]](https://thorax.bmj.com/content/thoraxjnl/73/6/519/DC5/embed/inline-supplementary-material-5.jpg?download=true){kind=link}
Supplemental material
![[SP6.jpg]](https://thorax.bmj.com/content/thoraxjnl/73/6/519/DC6/embed/inline-supplementary-material-6.jpg?download=true){kind=link}
Acknowledgments
We would like to thank the personnel of the Department of Pathology at the General University Hospital of Valencia and the animal housing facilities of the University of Valencia, Spain.
References
Footnotes
JM and BB contributed equally.
Contributors Conception and design: JM, BB, AM, JLO, FP-V, AC and JC. Data acquisition: JM, JE, EP, EA, EF, AC, EM and JC. Analysis and interpretation: all authors.
Funding This work was supported by the grants SAF2014-55322-P (JC), SAF2011-28150 (FPV) and SAF2014-55399-R, FIS PI14/01733 (JM), FIS PI17/02158 (JM), SAF2015-65368-R (EM), CIBERES (CB06/06/0027), TRACE (TRA2009-0311; Spanish Government) and by research grants from the Regional Government Prometeo II/2013/014 (JC, EM and JM) and ACIF/2016/341 (BB) from ‘Generalitat Valenciana’.
Disclaimer Funding entities did not contribute to the study design or data collection, analysis and interpretation nor to the writing of the manuscript.
Competing interests None declared.
Patient consent Obtained.
Ethics approval This study has been approved by the ethics committee of the University General Hospital of Valencia, Spain (CEIC22/2013).
Provenance and peer review Not commissioned; externally peer reviewed.