Article Text

Abstract
Background Idiopathic pulmonary fibrosis (IPF) is a rapidly progressive, fatal lung disease that affects older adults. One of the detrimental natural histories of IPF is acute exacerbation of IPF (AE-IPF), of which bacterial infection is reported to play an important role. However, the mechanism by which bacterial infection modulates the fibrotic response remains unclear.
Objectives Altered glucose metabolism has been implicated in the pathogenesis of fibrotic lung diseases. We have previously demonstrated that glucose transporter 1 (GLUT1)-dependent glycolysis regulates fibrogenesis in a murine fibrosis model. To expand on these findings, we hypothesised that GLUT1-dependent glycolysis regulates acute exacerbation of lung fibrogenesis during bacterial infection via AIM2 inflammasome activation.
Results In our current study, using a murine model of Streptococcus pneumoniae (S. pneumoniae) infection, we investigated the potential role of GLUT1 on mediating fibrotic responses to an acute exacerbation during bleomycin-induced fibrosis. The results of our current study illustrate that GLUT1 deficiency ameliorates S. pneumoniae-mediated exacerbation of lung fibrosis (wild type (WT)/phosphate buffered saline (PBS), n=3; WT/S. pneumoniae, n=3; WT/Bleomycin, n=5 ; WT/Bleomycin+S. pneumoniae, n=7; LysM-Cre-Glut1fl/f /PBS, n=3; LysM-Cre-Glut1fl/fl /S. pneumoniae, n=3; LysM-Cre-Glut1fl/fl /Bleomycin, n=6; LysM-Cre-Glut1fl/fl /Bleomycin+S. pneumoniae, n=9, p=0.041). Further, the AIM2 inflammasome, a multiprotein complex essential for sensing cytosolic bacterial DNA as a danger signal, is an important regulator of this GLUT1-mediated fibrosis and genetic deficiency of AIM2 reduced bleomycin-induced fibrosis after S. pneumoniae infection (WT/PBS, n=6; WT/Bleomycin+S. pneumoniae, n=15; Aim2−/−/PBS, n=6, Aim2−/−/Bleomycin+S. pneumoniae, n=11, p=0.034). GLUT1 deficiency reduced expression and function of the AIM2 inflammasome, and AIM2-deficient mice showed substantial reduction of lung fibrosis after S. pneumoniae infection.
Conclusion Our results demonstrate that GLUT1-dependent glycolysis promotes exacerbation of lung fibrogenesis during S. pneumoniae infection via AIM2 inflammasome activation.
- idiopathic pulmonary fibrosis
- pneumonia
- interstitial fibrosis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is the key question?
What is the mechanism by which superimposed Streptococcal pneumoniae infection regulates exacerbation of pulmonary fibrosis?
What is the bottom line?
Using in vivo and in vitro models of lung fibrosis, we illustrated how enhanced glucose transporter 1 (GLUT1)-dependent glycolysis can regulate exacerbation of pulmonary fibrosis during streptococcal infection via AIM2 inflammasome activation.
Why read on?
This is the first study to show a novel mechanism in which AIM2 inflammasome activation is a pivotal link between GLUT1-mediated glycolysis and exacerbation of lung fibrogenesis during bacterial infection.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a rapidly progressive, fatal lung disease that has a mean survival of less than 3 years after diagnosis.1 2 While the aetiology of IPF still remains unknown, several factors, such as genetic changes, infection, inhalation of fibres and particles (cigarette smoke and asbestos), and gastro-oesophageal reflux, have been implicated as causative agents in irreversible fibroproliferative disease of the lung.3 A detrimental natural history of IPF is the acute exacerbation of IPF (AE-IPF), a sudden acceleration of the disease or an idiopathic acute lung injury superimposed on pre-existing disease that leads to a significant fibrosis and decline in lung function.4 Although the dominant trigger of AE-IPF is unclear, there is some evidence to suggest a role for bacterial infection in its pathogenesis.5
Recent work has demonstrated that a distinct microbiota is present in both healthy and diseased airways.5–8 Using 16S rRNA gene sequencing, Han et al showed distinct alteration of microbiota in bronchoalveolar lavage (BAL) of patients with IPF, with Streptococcus and Staphylococcus species significantly associated with IPF progression.6 Additional studies, by Molyneaux et al, have reported Haemophilus, Streptococcus, Neisseria and Veillonella spp to be more abundant in BAL of patients with IPF.5 Taken together, these independent observations suggest that the presence of bacteria in the respiratory tract may play a role in driving IPF disease progression.
Glucose transporter 1 (GLUT1) is the most highly conserved and widely distributed glucose transporter in mammalian cells.9 10 Our previous findings demonstrated that GLUT1-dependent glycolysis is critical for parenchymal fibrosis and airway inflammation in a bleomycin-induced lung injury model.11 On a cellular level, heightened anaerobic glycolysis in macrophages is thought to mediate innate immune responses by activating inflammasomes.12 13 Inflammasomes are multiprotein complexes that recognise pathogen-associated molecular patterns and danger-associated molecular patterns, and ultimately promote the maturation and secretion of proinflammatory cytokines such as interleukin (IL)-1β and IL-18 via caspase-1 activation.14 Previous work has illustrated that NLRP3 inflammasome activation can contribute to lung fibrogenesis.15 16 However, in NLRP3-deficient mice, despite a dramatic decrease in bleomycin-induced pulmonary fibrosis, a significant amount of fibrosis remained.15 The AIM2 (Absent in Melanoma 2) inflammasome recognises self and foreign double-stranded DNA (dsDNA).17–21 Recent work has illustrated that extracellular mitochondrial DNA (mtDNA) concentrations are increased in plasma and BAL collected from patients with IPF, with excessive mtDNA levels in plasma being a predictor of mortality.22 As circulating cell-free mtDNA can contribute to AIM2 inflammasome-mediated responses in other models of chronic inflammation, the goal of our current study was to examine the role the AIM2 inflammasome plays in mediating acute exacerbation and lung fibrogenesis.23
In this study, we report that (1) GLUT1-dependent glycolysis mediates acute exacerbation of pulmonary fibrosis in response to streptococcal infection, (2) GLUT1-dependent glycolysis regulates AIM2 inflammasome activation and (3) genetic deficiency of AIM2 ameliorates exacerbation of pulmonary fibrosis in response to streptococcal infection. These findings support our overarching hypothesis that GLUT1-dependent glycolysis regulates acute exacerbation of lung fibrogenesis during bacterial infection via AIM2 inflammasome activation.
Materials and methods
Detailed methods are described in the online supplementary materials and methods.
Supplemental material
Animal studies
Bleomycin instillation: Young mice (8 weeks of age) were exposed to PBS or bleomycin (0.01 mg/mouse) via oropharyngeal aspiration. Streptococcus pneumoniae infection: All mice (8 weeks of age) underwent intranasal instillation with S. pneumoniae (ATCC 6303). For details, see online supplementary materials and methods.
Human lung samples
The Brigham and Women’s Hospital and Weill Cornell Medicine institutional review board (IRB) approved all experimental procedures involving use of human samples. Fresh-frozen human lung tissues were collected from patients with IPF undergoing lung transplantation or from failed donor controls. The human tissues provided by Brigham and Women’s Hospital are not identifiable. Therefore, neither IRB approval nor a notice of exemption is required for this project.
Glucose uptake assay
18F-fluorodeoxyglucose (18F-FDG) was administered intravenously via the tail vein. 18F-FDG injection and PET/CT imaging was performed using the Siemens Inveon system, followed by a CT scan. For details, see online supplementary materials and methods.
Transfection of Glut1 siRNA
Glut1 small interfering RNA (siRNA) was complexed with GenMute siRNA transfection reagent for 15 min before being added to macrophage cultures. Glut1 siRNA-transfected cells were cultured for 24 hours before Lipopolysaccharide (LPS) and/or poly(dA:dT) stimulation. For details, see online supplementary materials and methods.
Glycolytic function assay
Extracellular acidification rate (ECAR) as a parameter of glycolytic flux was measured on a Seahorse XF96 bioanalyser using the XF Glycolysis Stress Test kit according to the manufacturer’s instructions. For details, see online supplementary materials and methods.
ASC speck formation
Bone marrow–derived macrophages (BMDMs) transfected with non-target siRNA or siRNA for Glut1 and treated with vehicle or phloretin were seeded on chamber slides. For details, see online supplementary materials and methods.
Statistics
Error bars represent 95% CI as indicated in figure legends. Normality of data was assessed using the D’Agostino–Pearson omnibus normality test. Normally distributed data are for significance by a Student’s t-test or analysis of variance (ANOVA), as appropriate. Data that were not normally distributed were assessed for significance using a Kruskal-Wallis test followed by a Dunn post hoc test for multiple comparisons or Mann-Whitney U test for two group comparison using GraphPad Prism V.7.0. P<0.05 was considered statistically significant.
Results
S. pneumoniae infection exacerbates bleomycin-induced lung fibrosis
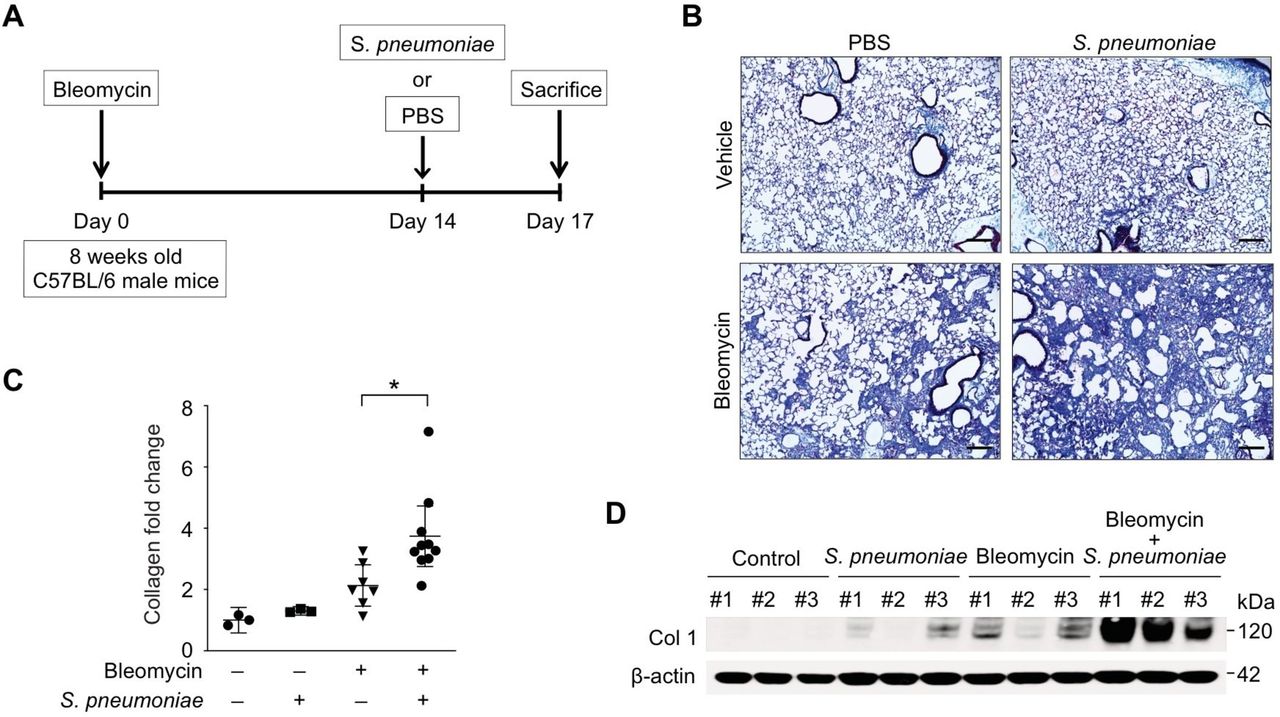
Patients with IPF are more susceptible to bacterial infection compared with healthy populations.24 25 Further, increased bacterial burden in BAL has been reported in patients with IPF who have rapid disease progression.6–8 Interestingly, among all infectious organisms screened in a recent study, streptococcal species were found to be the most abundant in patients with IPF whose disease process deteriorated rapidly.6 To examine the role of streptococcal infection on exacerbation of lung fibrosis, mice were initially instilled with bleomycin sulfate (0.01 mg/mouse; equivalent to 2 international units/kg, once, oropharyngeal) to induce injury and development of fibrosis (figure 1A). On day 14, a time point by which lung has developed significant fibrotic changes, mice were instilled with a highly virulent type 3 strain of S. pneumoniae (1×105 CFU) commonly associated with an increased relative risk of death26 27 (figure 1A). When compared with bleomycin or S. pneumoniae alone, mice treated with both bleomycin and S. pneumoniae showed a significant increase in fibrosis and collagen deposition in lung tissues (figure 1B). Measurement of collagen levels by Sircol assay illustrated that the level of acid-soluble collagen was elevated in lung tissue of bleomycin-treated mice, with significantly higher levels present after S. pneumoniae infection (figure 1C). Similarly, when compared with bleomycin treatment alone, collagen protein expression in lung was further augmented in response to streptococcal infection (Figure 1D, online supplementary figure 1A). In addition to augmented fibrosis, when compared with bleomycin or S. pneumoniae instillation alone, there was a significant increase in morbidity and dose-dependent mortality in bleomycin-treated mice infected with S. pneumoniae (online supplementary figure 1B and C).
Supplemental material
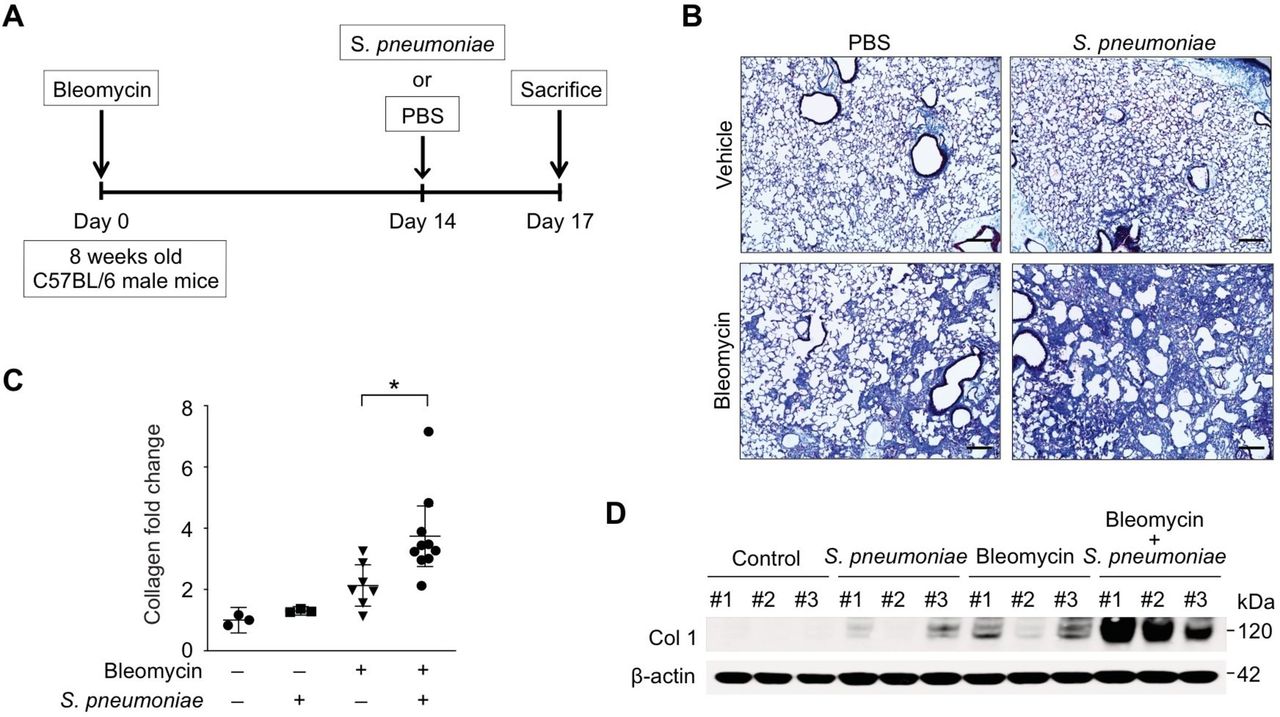
Streptococcal pneumoniae infection exacerbates bleomycin-induced lung fibrosis. (A) Experimental layout of primary bleomycin-induced lung injury (day 0 instillation of bleomycin 0.01 mg/mouse followed by day 14 instillation of PBS) and S. pneumoniae infection (day 0 instillation of bleomycin 0.01 mg/mouse followed by day 14 instillation of 1×105 CFU of S. pneumoniae). (B) Representative lung sections of bleomycin-treated mice subsequently infected with S. pneumoniae. Stained with Masson trichrome staining. Scale bars 200 μm. (C) Total lung collagen was quantified by Sircol assay (PBS/PBS, n=3; PBS/S. pneumoniae, n=3; bleomycin/PBS, n=14, bleomycin/S. pneumoniae, n=14). Fold change is relative to control lungs. Data are mean±95% Cl. *p<0.05 by analysis of variance. (D) Immunoblot analysis for collagen type 1 in bleomycin-treated lung tissue lysates in response to S. pneumoniae infection. β-actin served as the standard. Results are representative of three or more independent experiments.
To investigate if the observed exacerbation of fibrosis is specific to S. pneumoniae, we next examined the impact of additional pathogens, such as serotype 1 strain of S. pneumoniae and influenza A virus (H1N1, 1×106 PFU), on collagen production in bleomycin-treated lung. Notably, infection with serotype 1 strain of S. pneumoniae or H1N1 influenza did not increase collagen levels in bleomycin-treated mice (online supplementary figure 1D and E). We previously demonstrated that influenza caused mortality in aged mice but not in young mice.28 Consistent with our previous report, no lethality was observed in bleomycin-treated young mice after 1×106 PFU of H1N1 infection. These results suggest that fibrosis exacerbation in bleomycin-treated lung may be a unique characteristic of an infection with a serotype 3 strain of S. pneumoniae.
GLUT1-dependent glycolysis is increased in bleomycin-treated mice after S. pneumoniae infection
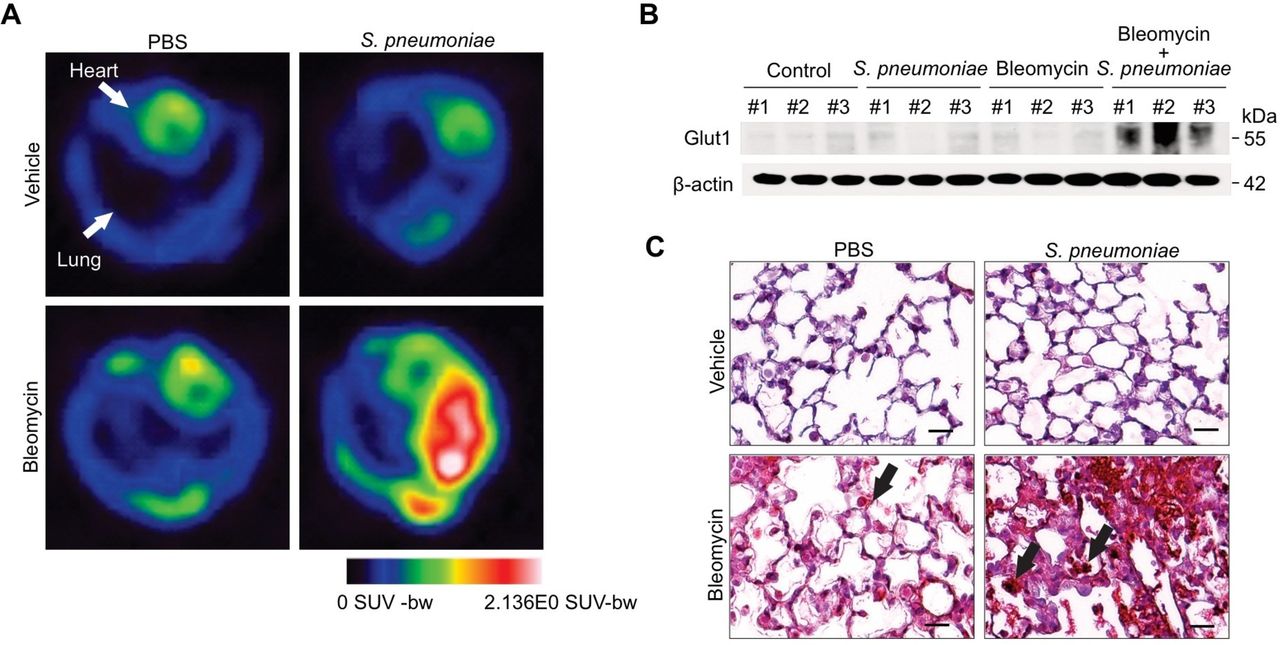
Our previous work illustrates that GLUT1-dependent glycolysis regulates lung fibrosis in response to bleomycin-induced lung injury.11 To expand on these findings, we investigated the effects of S. pneumoniae infection on GLUT1-dependent glycolysis in bleomycin-induced fibrosis. To demonstrate glycolytic activities in vivo, we used 18F-FDG-positron emission tomography scan (18F-FDG-PET), a non-invasive measure of cellular glucose metabolism. Our results illustrate that pulmonary uptake of 18F-FDG is highly intensified in bleomycin-treated mice in response to S. pneumoniae (figure 2A). As glycolysis in bleomycin-treated lung is augmented by S. pneumoniae, we next examined whether alterations in GLUT1 expression may underlie this metabolic change. To this extent, we analysed GLUT1 protein levels in bleomycin-treated lung tissue after S. pneumoniae instillation. When compared with bleomycin alone, GLUT1 expression was significantly increased in bleomycin-treated lung in response to streptococcal infection (figure 2B and C, online supplementary figure 2). Further, this augmentation of GLUT1 expression was seen predominantly in inflammatory cells (figure 2C).

GLUT1-dependent glycolysis is increased in bleomycin-treated mice after Streptococcal pneumoniae infection. (A) 18F-FDG-PET scan of lungs from primary bleomycin-induced lung injury (day 0 instillation of bleomycin followed by day 14 instillation of PBS) and S. pneumoniae infection 6 (day 0 instillation of bleomycin followed by day 14 instillation of 1×106 CFU of S. pneumoniae). 18F-FDG uptake is more in bleomycin-treated lung in response to S.pneumoniae infection compared with controls. (B) lmmunoblot analysis for GLUT1 in bleomycin-treated lung tissue lysates in response to S. pneumoniae infection. β-actin served as the standard. (C) Immunohistochemical staining of GLUT1 (red) in bleomycin-treated lung reveals enhanced signal in fibrotic foci and inflammatory cells (solid arrow). Scale bars 200 µm. Results are representative of three or more independent experiments. GLUT1, glucose transporter 1; 18F-FDG-PET, 18F-FDG-positron emission tomography.
GLUT1-dependent glycolysis regulates exacerbation of lung fibrosis after S. pneumoniae infection
We next investigated if GLUT1-dependent glycolysis regulates the exacerbation of lung fibrosis in bleomycin-treated mice during superimposed S. pneumoniae infection. We generated myeloid cell-specific Glut1 knockout (LysM-Cre-Glut1 fl/fl ) mice by crossing Glut1 floxed mice (Glut1 fl/fl) with LysM-Cre mice in which Cre recombinase is expressed in macrophages, monocytes and dendritic cells. GLUT1 expression was augmented in lung tissue isolated from wild-type (WT) mice after bleomycin treatment and subsequent infection on day 14 with S. pneumoniae (Figure 3A, online supplementary figure 3A). Myeloid cell-specific Glut1 deficiency led to decreased protein levels of collagen in bleomycin-treated lung in response to a S. pneumoniae infection (figure 3A, online supplementary figure 3A). Further, acid-soluble collagen levels in bleomycin-treated lung in response to infection were significantly decreased in LysM-Cre-Glut1fl/fl mice (figure 3B). Histologically, when compared with the WT mice, LysM-Cre-Glut1fl/fl mice showed reduced accumulation of collagen in the subepithelial area and interalveolar septum (figure 3C). Clinically, LysM-Cre-Glut1fl/fl mice had less morbidity compared with WT mice after superimposed S. pneumoniae infection (online supplementary figure 3B). The impact of GLUT1 deficiency on glycolysis was examined using 18F-FDG-PET scans. Pulmonary uptake of 18F-FDG was found to be significantly reduced in bleomycin-treated LysM-Cre-Glut1fl/fl mice in response to S. pneumoniae (figure 3D). In sum, these results suggest that GLUT1-dependent glycolysis regulates exacerbation of fibrosis in response to S. pneumoniae infection.

GLUT1-dependent glycolysis regulates exacerbation of lung fibrosis after Streptococcal pneumoniae infection. (A) Immunoblot analysis for Glut1 and collagen type 1 in bleomycin-treated WT and LysM-Cre-Glutimmicefl/fl after S. pneumoniae infection. β-actin served as the standard. (B) Total lung collagen was quantified by Sircol assay (WT/PBS, n = 3; WT/S. pneumoniae, n = 3, WT/Bleomycin n = 5, WT/Bleomycin+S . pneumoniae, n = 6; LysM-Cre-Glutlfl/fl/PBS, n = 3; LysM-Cre-Glut1fl/fl /S. pneumoniae, n = 3; LysM-Cre-Giutr1 fl/ flBleomycin, n=6; LysM-Cre-G/ut1fl/fl /Bleomycin+S . pneumoniae, n=6). Fold change is relative to PBS-treated WT lungs. Data are mean±95% Cl. *p<0.05 by ANOVA. (C) Representative lung sections of WT and LysM-Cre-Glut 1 fl/fl mice after bleomycin treatment followed by S. pneumoniae infection. Stained with Masson trichrome staining. Scale bars 200 µm. (D) 18F-FDG-PET scan showed that FDG uptake is less in LysM-Cre-Glut 1 fl/fl mice after bleomycin treatment followed by S. pneumoniae infection. Results are representative of two or more independent experiments.18F-FDG-PET, 18F-FDG-positron emission tomography; WT, wild type.
AIM2 inflammasome expression and activation is augmented in bleomycin-treated lung after S. pneumoniae infection
Although the role of inflammation in fibrosis has engendered debate, there is evidence that epithelial injury and cell death lead to chronic inflammation and fibrosis.29–31 In agreement with these findings, our results illustrate a significant increase in the total number of inflammatory cells in BAL in response to bleomycin alone, which was further augmented by S. pneumoniae (figure 4A). Of note, while there are similar number of macrophages in BAL in bleomycin and bleomycin+S. pneumoniae groups, there was a significant increase in the number of neutrophils present in the BAL collected from bleomycin+S. pneumoniae group when compared with bleomycin alone (online supplementary figure 4A). Given the impact of overly heightened inflammasome expression and activation on fibrosis, we next investigated the role of inflammasomes on fibrosis exacerbation in response to S. pneumoniae. Specifically, we examined if AIM2 inflammasome expression in bleomycin-treated lung was altered in response to infection with S. pneumoniae. Immunohistologically, we found AIM2 expression to be increased in alveolar macrophages (solid arrows) after treatment with either bleomycin or S. pneumoniae alone, and further heightened in a bleomycin-treated lung in response to streptococcal infection (figure 4B). Consistent with these results, immunoblot analysis showed that AIM2 protein levels, caspase-1 activation and IL-1β cleavage in lung tissues of bleomycin-treated mice were augmented after S. pneumoniae infection (Figure 4C, Supplementary figure 4B). IL-1β and IL-18 production in lung tissue was heightened in bleomycin-treated mice in response to streptococcal infection (figure 4D,E). Since the NLRP3 inflammasome is a potent mediator of lung fibrosis, we next examined whether AIM2 inflammasome activation regulates lung fibrosis exacerbation in NLRP3 knockout (Nlrp3 −/− ) mice. Consistent with WT mice, Nlrp3 −/− mice treated with bleomycin and subsequently infected with S. pneumoniae showed a significant increase in collagen production in lung tissue when compared with treatment with bleomycin or S. pneumoniae alone (online supplementary figure 4C). In addition, in the absence of NLRP3, the inflammatory cell count in BAL was also significantly increased in bleomycin-treated lung in response to S. pneumoniae (online supplementary figure 4D). Further, caspase-1 activation and IL-1β cleavage in lung tissues were increased in bleomycin-treated lung mice in response to S. pneumoniae, despite the absence of NLRP3 (online supplementary figure 4E). To expand these findings, we next examined if AIM2 expression was increased in human IPF lung tissue. Immunoblot analysis showed that levels of AIM2 expression were significantly elevated in advanced IPF lungs compared with control lungs, whereas levels of NLRP3 expression were comparable between IPF lungs and controls (online supplementary figure 5A and B).

AIM2 inflammasome expression and activation is augmented in bleomycin-treated lung after Streptococcal pneumoniae infection. (A) Total cell count in bronchoalveolar lavage (BAL) in bleomycin-treated mice 3 days after S. pneumoniae infection. Data are mean±95% Cl. *p<0.05. (B) Immunohistochemical staining of AIM2 (red) in bleomycin-treated lung reveals enhanced signal in inflammatory cells (solid arrow). Scale bars 200 µm. (C) lmmunoblot analysis for AIM2, activated caspase-1, cleaved IL-1β (black arrows) in bleomycin-treated lung after S. pneumoniae infection.(β-actin served as the standard). Amounts of IL-1β (D) and IL-18 (E), as determined by ELISA in lung tissues (50 µg) after S. pneumoniae infection for 3 days (PBS/PBS, n=3; PBS/S. pneumoniae, n=3; Bleomycin/PBS, n=8, Bleomycin/S. pneumoniae, n=8). Data are mean±95% Cl. *p<0.05. Results are representative of two or more independent experiments. BAL, bronchoalveolar lavage; IL, interleukin.
GLUT1-dependent glycolysis regulates AIM2 inflammasome activation
We next investigated whether GLUT1-dependent glycolysis can regulate inflammation by modulating AIM2 inflammasome activation during S. pneumoniae infection. To this extent, we first examined whether GLUT1 regulates glycolysis in BMDMs from Nlrp3 −/− mice. We treated primary BMDMs with missense or GLUT1-specific siRNA and measured glycolic activity using Seahorse analysis. We assessed the ECAR, as a measure of lactate production (a surrogate for the glycolytic rate), and ECAR was measured after the addition of glucose. Oligomycin (2 µM) was injected after glucose (10 mM) to inhibit mitochondrial respiration. 2DG (100 mM) injection reduced ECAR down to baseline by inhibiting hexokinase. With Glut1 knockdown in macrophages, a significant decrease in ECAR after glucose challenge was observed (figure 5A). We then used LPS priming followed by poly(dA:dT) transfection, a potent AIM2 inflammasome-specific activator,20 to test if GLUT1-dependent glycolysis regulates AIM2 inflammasome activation. Poly(dA:dT) is a synthetic analogue of microbial dsDNA that has been used to mimic intracellular bacteria and dsDNA virus infection which is known to activate AIM2 inflammasome. When Glut1 expression was knocked down in BMDMs, a significant decrease in caspase-1 activation and IL-1β cleavage after LPS and poly (dA:dT) stimulation was observed when compared with missense siRNA-treated BMDMs (figure 5B,C). GLUT1-dependent glycolysis was furthermore found to regulate the oligomerisation of ASC, which is required for AIM2 inflammasome formation and AIM2-dependent caspase-1 activation. The formation of ASC specks induced by LPS and poly(dA:dT) stimulation was significantly increased in WT BMDMs when compared with similarly treated Glut1 knockdown BMDMs (figure 5D,E).

GLUT1-dependent glycolysis regulates AIM2 inflammasome activation in vitro. (A) Extracellular acidification rate (ECAR) was measured in BMDMs from Nlrp3−/ − mice (BMDMs, 1×105 cells/well) transfected with non-target siRNA (control siRNA) or siRNA for Glut1 24 hours prior to LPS and poly(dA:dT) stimulation, a potent AIM2 inflammasome activator. Data are mean±SEM. (B) lmmunoblot analysis for activated caspase-1, cleaved IL-1β (black arrows) in BMDMs transfected with control siRNA or siRNA for Glut1 24 hours prior to LPS and poly(dA:dT) stimulation. β-actin served as the standard. (C) Quantification of IL-1β levels from BMDMs from Nlrp3−/ − mice transfected with control siRNA or siRNA for Glut1 24 hours prior to LPS and poly(dA:dT) stimulation. Representative immunofluorescence images (n=5 individual images per group) of ASC speck formation (white arrows) images (D) and quantification (E) in BMDMs from Nlrp3−/ − mice transfected with control siRNA or siRNA for Glut1 that were stimulated without or with LPS and poly(dA:dT). Scale bars, 20 µm. Throughout, data are mean±95% Cl. *p<0.05, **p<0.01 by analysis of variance. Results are representative of three or more independent experiments. BMDMs, bone marrow–derived macrophages; IL, interleukin; GLUT1, glucose transporter 1.
Pharmacological inhibition of GLUT1 in BMDMs was then performed and responses to LPS and poly(dA:dT) were assessed. GLUT1 activity in BMDMs was inhibited by treatment with phloretin, a potent GLUT1 inhibitor. Similar to our GLUT1 siRNA findings, phloretin suppressed ECAR after glucose challenge (online supplementary figure 6A). Moreover, caspase-1 activation and IL-1β production were decreased after LPS and poly (dT:dA) stimulation when compared with vehicle control (online supplementary figure 6B and C). ASC oligomerisation was also significantly reduced in phloretin-treated BMDMs as compared with untreated BMDMs, as assessed by the number of cells with ASC specks that were induced by LPS and poly(dA:dT) (online supplementary figure 6D and E).
We next examined if GLUT1 deficiency can reduce AIM2 inflammasome activation and lung inflammation in our murine fibrosis exacerbation model. The total number of inflammatory cells present in the BAL was significantly decreased in bleomycin-treated LysM-Cre-Glut1fl/fl mice after S. pneumoniae infection (Figure 6A, online supplementary figure 7A). When compared with WT mice, AIM2 expression, caspase-1 activation and production of IL-1β and IL-18 were all decreased in lung isolated from bleomycin Glut1 knockout mice in response to S. pneumoniae (figure 6B–D). These results suggest that GLUT1-dependent glycolysis is critical for inflammation and the activation of AIM2 inflammation in vitro and in vivo.
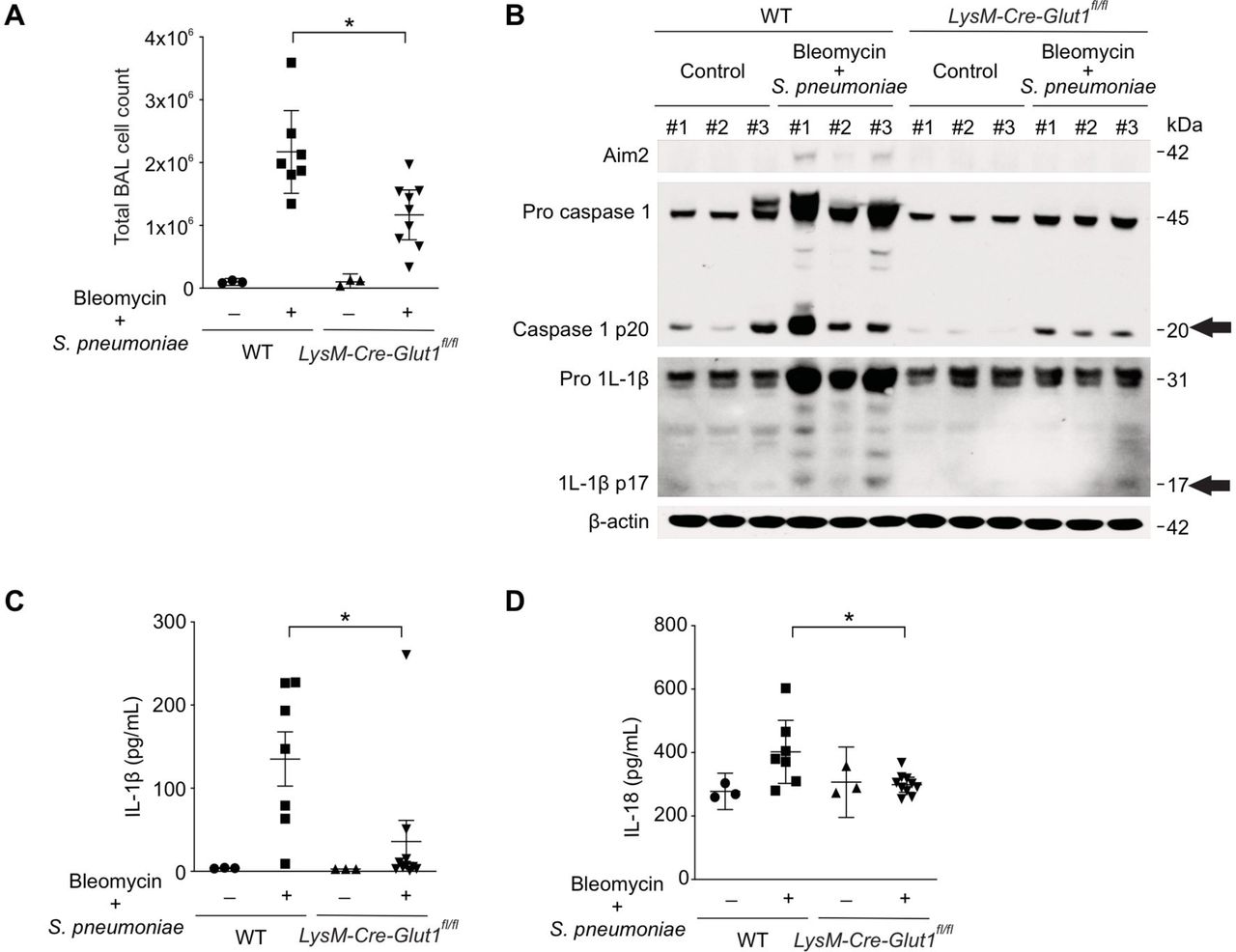
GLUT1-dependent glycolysis regulates AIM2 inflammasome activation in murine fibrosis exacerbation model. (A) Total BAL cell count in bleomycin-treated WT and LysM-Cre-Giut1fl/fl mice 3 days after Streptococcal pneumoniae infection. (B) lmmunoblot analysis for AIM2, activated caspase-1, cleaved IL-1β (black arrows) in bleomycin-treated lung after S. pneumoniae infection. β-actin served as the standard. Amounts of IL-1β (C), IL-18 (D), as determined by ELISA in lung tissues (50 µg) after S. pneumoniae infection for 3 days. (WT/PBS, n=3; WT/Bleomycin+S. pneumoniae, n=7; LysM-Cre-Giut1fl/fl// /PBS, n=8, LysM-Cre-Giut1fl/fl /Bieomycin+S. pneumoniae, n=1 0). Throughout, data are mean±95% Cl. *p<0.05 by analysis of variance. Results are representative of three or more independent experiments. BAL, bronchoalveolar lavage; IL, interleukin; GLUT1, glucose transporter 1; WT, wild type.
Deficiency of AIM2 ameliorates fibrosis in bleomycin-treated lung after S. pneumoniae infection
We next examined the role of the AIM2 inflammasome on the exacerbation of fibrosis observed after S. pneumoniae infection. When compared with WT, Aim2 knockout (Aim2 −/− ) mice had reduced accumulation of collagen in the subepithelial area and interalveolar septum in response to S. pneumoniae instillation 14 days after bleomycin treatment (figure 7A). Further, there was a reduction in the acid-soluble collagen content present in Aim2 −/− lung tissue in response to S. pneumoniae (figure 7B). As assessed by immunoblot analysis, lung tissue collected from Aim2 −/− mice exhibited decreased collagen protein levels compared with similarly treated WT mice (figure 7C, online supplementary figure 8A). Interestingly, there was similar collagen content in the lung of Aim2 −/− mice and WT lung after treatment with bleomycin alone (online supplementary figure 8B). Genetic deficiency of AIM2 ameliorated the profound weight loss seen in WT mice observed after bleomycin instillation and subsequent S. pneumoniae infection (p<0.0001 by one-way ANOVA and p=0.016 between WT/Bleomycin+S. pneumoniae and AIM2−/−/Bleomycin+S. pneumoniae by post hoc analysis) (online supplementary figure 8C). Consistent with our understanding that AIM2 functions by activating caspase-1 to promote cleavage of proinflammatory cytokines, we found that lung tissue of Aim2 −/− mice exhibited less caspase-1 activation and decreased levels of IL-1β and IL-18 (figure 7D–F). In sum, our findings demonstrate that GLUT1-dependent glycolysis promotes exacerbation of lung fibrogenesis during S. pneumoniae infection via AIM2 inflammasome activation (figure 7G).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Deficiency of AIM2 ameliorates fibrosis exacerbation in bleomycin-treated lung after Streptococcal pneumoniae infection. (A) Representative lung sections of WT and AIM2−/ − mice after bleomycin treatment followed by S. pneumoniae infection. Stained with Masson trichrome staining. Scale bars 200 µm. (B) Total lung collagen was quantified by Sircol assay (WT/PBS, n=6; WT/Bleomycin+S. pneumoniae, n=15; AIM2−/ −/PBS, n=6, AIM2/Bleomycin+S. pneumoniae, n=11). (C) Immunoblot analysis for collagen type 1 in bleomycin-treated WT and Aim2−/ − mice after S. pneumoniae infection. β-actin served as the standard. (D) Immunoblot analysis for AIM2, activated caspase-1, cleaved IL-1β (black arrows) in bleomycin-treated WT and AIM2−/ − mice after S. pneumoniae infection. β-actin served as the standard. Amounts of IL-β-β (E), IL-18 (F), as determined by ELISA in lung tissues (50 µg) after S. pneumoniae infection for 3 days (WT/PBS, n=3; WT/Bleomycin+S. pneumoniae, n=10; Aim2−/ −/PBS, n=3, Aim2−/ −/Bleomycin+S. pneumoniae, n=5). Throughout, data are mean±95% Cl. *p<0.05 by analysis of variance. Results are representative of three or more independent experiments. (G) Schematic of the proposed relationships between GLUT1-dependent glycolysis and AIM2 inflammasome activation-mediated fibrosis. IL, interleukin; GLUT1, glucose transporter 1; WT, wild type.
Discussion
In this study, we identify GLUT1-dependent glycolysis as a critical regulator of fibrogenesis in response to S. pneumoniae infection via AIM2 inflammasome activation. We have previously demonstrated that GLUT1-dependent glycolysis regulates fibrogenesis in murine lung via fibroblast activation. The novel finding in our current study is that GLUT1-dependent glycolysis in macrophages regulates fibrosis via AIM2 inflammasome activation during a subsequent infection with S. pneumoniae.
There have recently been multiple studies investigating the role of lung microbiome in pathogenesis of IPF. Previous studies using 16S rRNA gene sequencing have demonstrated that when compared with control subjects, patients with IPF have a higher bacterial load in BAL. In addition, differences in specific operational taxonomic units between IPF cases and control subjects have been reported, notably the presence of more abundant Streptococcus, Haemophilus, Neisseria and Veillonella species in patients with IPF.5 In another study, Martinez and colleagues have recently presented data for the abundance of streptococcal species in individuals with IPF, and strong correlation between streptococcal burden and IPF disease progression and survival.6 These independent observations suggest that the presence of treptococcal species, the most common pathogen causing pneumonia in patients with IPF, may play a role in driving IPF disease progression. Based on these association studies, we tested whether S. pneumoniae infection has mechanistic influence on the exacerbation of lung fibrosis. Prior work has suggested pneumolysin, a pore-forming toxin from S. pneumoniae, as a cause of lung fibrosis in murine models.32 However, mechanisms by which S. pneumoniae regulate inflammation and fibrogenesis in fibrosis exacerbation remain unclear. We are the first to report that secondary S. pneumoniae infection exacerbates lung fibrogenesis by augmenting glycolysis through GLUT1 upregulation. In agreement with previous work, our results also illustrated that influenza A (H1N1) does not exacerbate murine lung fibrosis.33 In agreement with these findings, we do not detect an increase in lung fibrosis in response to influenza A infection.
Glycolysis is the primary bioenergetic pathway in many human disease states.34 Studies have reported that glycolysis and its metabolites are associated with disease severity and progression in chronic lung diseases such as IPF.35–39 Lactate is the final metabolite of glycolytic pathway, and Kottmann et al showed that lactate production promotes lung fibroblasts differentiation and activation via a pH-dependent activation of transforming growth factor (TGF)-β.40 It has been suggested that, mechanistically, specific glycolytic enzymes such as 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) and pyruvate kinase muscle 2 (PKM2) provoke fibrogenesis by activating fibroblast activation and differentiation.41 We have previously reported that GLUT1, the most abundant glucose transporter in mammalian cells, is enhanced in aged mice treated with bleomycin.11 In our current study, we demonstrate that GLUT1 expression is further increased in bleomycin-treated lung in response to streptococcal infection. Functional analysis using 18F-FDG-PET, a non-invasive functional measure to detect glucose uptake, revealed 8F-FDG uptake to be more prominent in fibrotic lungs after infected with S. pneumoniae.
Our study illustrates that abolishing myeloid cell-specific GLUT1 ameliorates fibrogenesis exacerbated by S. pneumoniae infection in the bleomycin-induced mouse lung injury model. Our findings support prior evidence that activated macrophages contribute to the pathogenesis of pulmonary fibrotic process in IPF lungs and murine models.29 We have also uniquely demonstrated that inhibiting GLUT1-dependent glycolysis limits the capacity of macrophages to activate AIM2 inflammasome. Our findings suggest that inhibition of GLUT1-mediated glycolysis in macrophages is a promising therapeutic target for the treatment of chronic fibrotic lung diseases such as IPF.
The role of glycolysis on the regulation of inflammasome activation in immune cells, such as macrophages, has previously been established.12 13 Glycolysis mediated by PKM2, an enzyme that dephosphorylates phosphoenolpyruvate to pyruvate, promotes both NLRP3 and AIM2 inflammasome activation.13 Hexokinase 1, which phosphorylates glucose to produce glucose-6-phosphate (G6P), is another glycolytic enzyme reported to regulate NLRP3 inflammasome activation.12 Although these and other glycolytic enzymes have been reported to mediate activation of inflammasomes, the role of GLUT1 in inflammasome activation has not previously been investigated. In the current study, we show that AIM2 inflammasome activation is regulated by GLUT1-dependent glycolysis in vitro. Further, our results demonstrate that lungs of myeloid cell-specific Glut1 knockout mice exhibit decreased levels of AIM2 expression and activation in vivo.
As we previously reported NLRP3 inflammasome activation to play an important role in fibrogenesis in our murine fibrosis model15 and NLRP3 is reported to be regulated by glycolysis,12 we used BMDMs isolated from Nlrp3 knockout mice to eliminate the effect of crosstalk between AIM2 and NLRP3. Consistent with WT mice, our results demonstrated that Nlrp3 knockout mice still have a significant increase in fibrosis and inflammation in response to bleomycin treatment followed by S. pneumoniae infection. In addition, we used BMDMs isolated from Nlrp3 knockout mice to exclude the effect of NLRP3 inflammasome activation. Similar to WT BMDMs, Nlrp3 knockout BMDMs showed decreased AIM2 inflammasome activation by poly(dA:dT) stimulation after both genetic and pharmacological inhibition of GLUT1.
Our findings provide a novel mechanism in which AIM2 inflammasome activation is a pivotal link between GLUT1-mediated glycolysis and exacerbation of lung fibrogenesis during bacterial infection. GLUT1 has been a therapeutic target for many solid tumours.42 Our current study extends promise that GLUT1 may be used to monitor IPF disease severity and progression, and also serve as a potential target of therapy for exacerbation of fibrosis, particularly in the setting of bacterial infection.
Acknowledgments
We acknowledge members of the Brigham and Women’s Hospital Interstitial Lung Disease Program and its Lung Center Biorepository for providing human samples.
References
Footnotes
Contributors Conception and design: SJC, J-SM and HS-D. Analysis and interpretation: SJC, J-SM, KN, HSY, RH, HH and HS-D. Drafting the manuscript for important intellectual content: SJC, J-SM, AMKC and HS-D.
Funding This work was supported by the K08HL138285 (SJC), American Lung Association Biomedical Grant (SJC), R01AG052530 (HWS) and R01AG056699 (HWS).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All mouse experimental protocols were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College (protocol Nos. 2014-0059 and 2014-0052; Weill Cornell Medical College, New York, New York, USA).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.