Article Text

Abstract
Background: Rhinovirus infection is responsible for considerable morbidity and mortality as the major cause of exacerbations of asthma, and is also known to induce exacerbations of cystic fibrosis and chronic obstructive pulmonary disease. Exacerbations of these diseases are also frequently associated with bacterial and atypical bacterial infection. Alveolar macrophages are the major immune cells in the airways and are important in defence against bacterial infections.
Methods: The authors investigated whether rhinovirus modifies cytokine release, the pattern recognition receptor expression and phagocytosis by human alveolar macrophages in response to bacterial products.
Results: Viable rhinovirus was detected in macrophages up to 3 days after exposure and viral RNA expression persisted for 10 days. Infectious but not UV inactivated rhinovirus increased tumour necrosis factor α (TNFα) and interleukin (IL)8 release by macrophages. In contrast, infectious rhinovirus impaired lipopolysaccharide and lipoteichoic acid induced TNFα and IL8 secretion by macrophages. Rhinovirus induced impairment of macrophage antibacterial immune responses did not involve IL10, prostaglandin E2 or downregulation of Toll-like receptor 2. Furthermore, the macrophage phagocytic response to labelled bacterial particles, but not to latex beads, was impaired.
Conclusion: The authors have identified impairment of cytokine responses to bacterial lipopolysaccharide and lipoteichoic acid by alveolar macrophages in response to infectious rhinovirus. Virus induced impairment of antibacterial host defence has important implications in the pathogenesis of exacerbations of respiratory diseases.
Statistics from Altmetric.com
Acute exacerbations of the chronic respiratory disorders asthma, cystic fibrosis and chronic obstructive pulmonary disease (COPD) are the major cause of morbidity, mortality and health care costs related to these diseases. The pathogenesis of acute exacerbations is poorly understood and therefore a better assessment of the underlying mechanisms will help to develop new therapeutic strategies.
Viral respiratory tract infections are the major precipitants of asthma exacerbations in both children1–4 and adults.5–10 There is increasing evidence that exacerbations of COPD11 ,12 and cystic fibrosis13 are also induced by viral infections. Of the different virus types associated with exacerbations of each of these diseases, rhinoviruses (RV) account for approximately two-thirds of the viruses identified.4–7 10 14
Bacterial infection is also associated with the majority of exacerbations of COPD15 and cystic fibrosis.16 A recent study has identified viral and bacterial coinfection in one-quarter of COPD exacerbations, and reported that exacerbations with coinfection were of increased severity.17 Atypical bacteria have also been shown to be associated with exacerbations of COPD.18–20 The role of bacterial infections in asthma exacerbations is more controversial, but patients with asthma have recently been shown to have increased susceptibility to invasive bacterial infection,21 and importantly atypical bacterial infection was reactivated in virus induced asthma exacerbations22 and related to exacerbation frequency.23 Finally, we have recently shown that an antibiotic therapy active against atypical bacteria is effective in the treatment of asthma exacerbations.24
The possibility arises that virus induced exacerbations may be further worsened by the occurrence of a concomitant bacterial superinfection.25 The occurrence of bacterial superinfection as a consequence of influenza viral infection is well documented. Combined viral and bacterial infection is likely to be due in part to increased bacterial adherence to infected epithelial cells, as demonstrated in vitro for influenza virus,26 respiratory syncytial virus27 and RV.28 However, little else is known about the possibility of RV infection increasing the risk of bacterial infection.
RV infects the lower respiratory tract of infants29 and adults,30 with the major site of infection occurring in bronchial epithelial cells.30 The importance of epithelial cells in antiviral immunity has recently been shown in that epithelial cells from subjects with asthma have reduced RV induced production of interferon β, with consequent increased RV replication.31 However, leucocytes present in the airway are also important in host defence against infections. The predominant leucocyte found at this location is the alveolar macrophage and these have also been shown to be deficient in interferon production in asthma.32 In support of their antimicrobial role, macrophages produce inflammatory cytokines to recruit cells of the adaptive immune system, express a number of innate pattern recognition receptors (PRRs) capable of detecting bacterial products, and phagocytose bacterial organisms. In addition, we have recently shown that the production of TNFα by macrophages in response to RV requires viral replication.33
RV has been shown to bind, enter and activate alveolar macrophages, although productive replication was not demonstrated.34 In this study, we investigated the hypothesis that RV infection of alveolar macrophages downregulates their antibacterial responses, thereby increasing the host’s susceptibility to bacterial infections.
To test this hypothesis, in the absence of the existence of a small animal model of RV infection, isolated human alveolar macrophages exposed to either infectious or UV inactivated RV were stimulated with the gram negative and gram positive bacterial products lipopolysaccharide (LPS) or lipoteichoic acid (LTA) and the production of proinflammatory cytokines tumour necrosis factor α (TNFα) and interleukin (IL)8, PRR expression and phagocytic ability was assessed.
MATERIALS AND METHODS
For full details see supplementary material online.
Isolation and in vitro culture of alveolar macrophages
Alveolar macrophages were isolated from resected lung tissue by parenchymal lavage, and plated in 10% FCS in RPMI medium (5×105/ml). Following washing, adherent cells (ie, alveolar macrophages) from each subject were cultured in the presence of medium alone, or infected/stimulated with RV, bacterial LPS and LTA (Sigma, Australia).
RV propagation and titration
Stocks of human RV-16 and human RV-2 were amplified by growth in Ohio HeLa cells and UV inactivated (UVi), as previously described.35 ,36 Following exposure to RV, virion production was assessed by titration assay35 and RT-PCR.37
RV exposure and toxin stimulation of alveolar macrophages
Alveolar macrophages were exposed to RV at a multiplicity of infection (MOI) of 0.1 or 1 at 37°C for 1 h. To determine bacterial toxin responsiveness to LPS, infected cells were stimulated for 24 h with LPS derived from Escherichia coli (10 ng/ml) at either 1 or 4 days post infection or LTA derived from Staphylococcus aureus (10 ng/ml) at 1 day post infection.
ELISA
ELISA kits for eotaxin, TNFα, IL8, IL10 and interferon γ (IFNγ) were purchased from R&D Systems Europe (Abingdon, UK). ELISAs were carried out according to the manufacturer’s instructions. The detection limits of these assays are 15.6 pg/ml for all except IL10 (33 pg/ml) and eotaxin (25 pg/ml).
Cell labelling for flow cytometry
Antibody binding and subsequent flow cytometric analysis was performed to assess the cell surface expression of Toll-like receptor 2 (TLR2) (Santa Cruz, California, USA), TLR4 (Santa Cruz) and CD14 (BD, North Ryde, Australia). Appropriate isotype controls were purchased from BD (North Ryde, Australia). Briefly, alveolar macrophages were detached by scraping, resuspended in calcium and magnesium free PBS/FBS and incubated with the antibodies for 1 h at 4°C. The cells were washed twice and resuspended in PBS/FBS prior to analysis.
Phagocytosis assay
Phagocytosis of labelled bacterial particles was assessed using the Vibrant Phagocytosis Assay Kit (Invitrogen, Australia) according to the manufacturer’s instructions. Phagocytosis of latex beads (Polysciences 0.5 μM Fluoresbrite Microparticles; Polysciences, Inc., Warrington, USA) was determined using flow cytometry.
Flow cytometry
Fluorescence was analysed by FACScan flow cytometry (Becton Dickinson, San Jose, California, USA). Macrophages (10 000 events) were acquired by gating on forward and side angle scatter properties.
Statistical methods and analysis of results
For experiments in which measurements were compared between the constitutive and experimental response observed in cells from the same donor, repeated measures ANOVA with Dunnett’s post test or paired Student’s t test was used. Post hoc tests were carried out only on data tables that were shown to be significantly different by ANOVA. Data were analysed using GraphPad Prism V.4.00 for Windows (GraphPad Software, San Diego, California, USA). A probability level of 95% (p⩽0.05) was considered as the threshold for statistical significance.
RESULTS
Patient demographics
Alveolar macrophages were isolated from 46 subjects: 31 males (mean age 63 (SD 11.6) years) and 15 females (59.9 (14.4)). The clinical diagnoses were lung cancer in 32 (69.6%), other thoracic malignancies in nine (19.6%), interstitial lung disease in one (2%), emphysema in one (2%) and unknown in three (6.5%).
Replication of RV in human alveolar macrophages
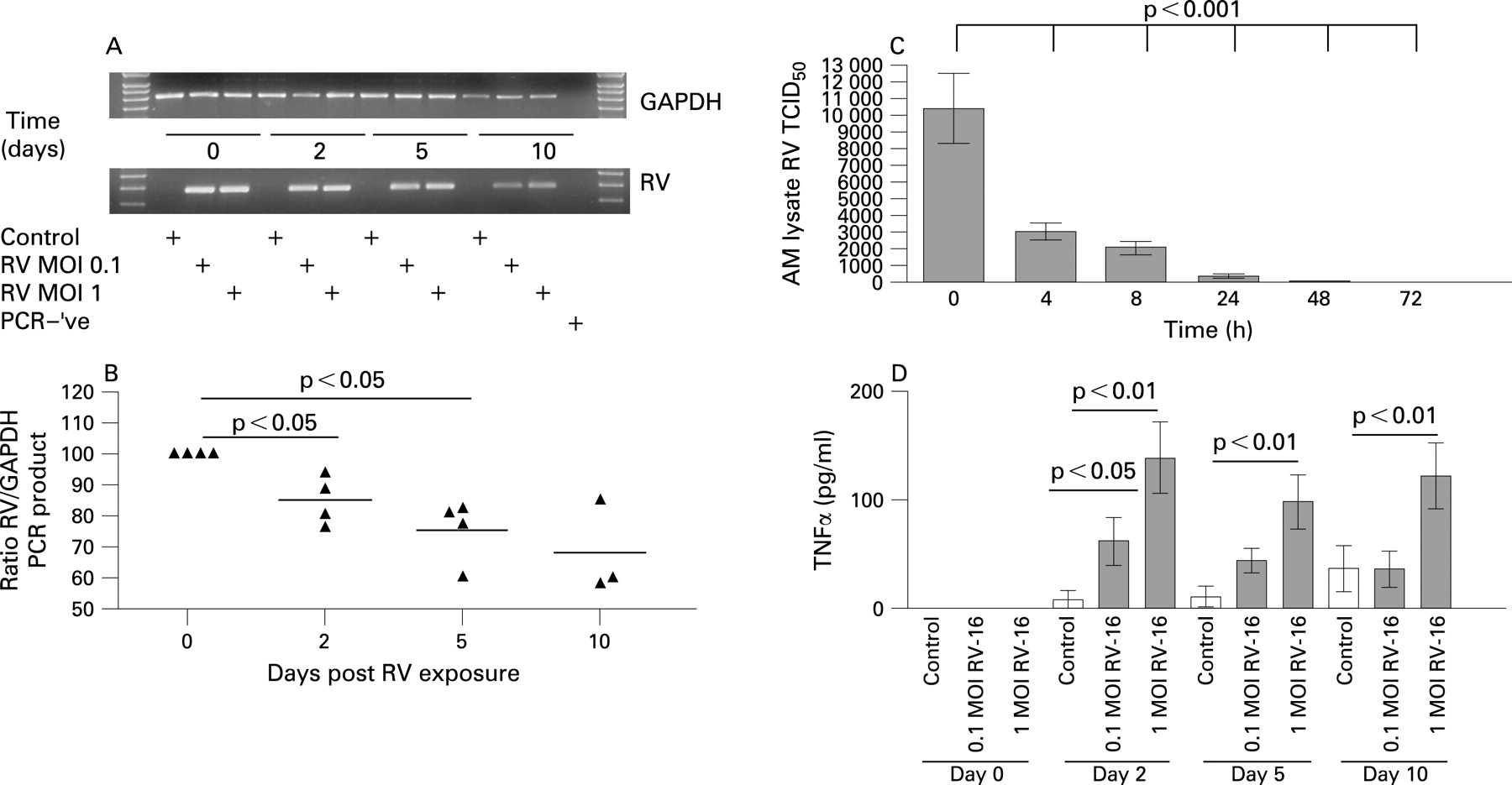
RV PCR and titration assays were used to examine if RV was capable of replicating within alveolar macrophages. In alveolar macrophages infected with an initial minimal virus load of 0.1 or 1 (data not shown) MOI RV, RV RNA was detected up to 10 days post infection (fig 1A) and was significantly reduced at 2 days post infection (fig 1B), with further reductions at either 5 or 10 days. To examine if the persistence of infectious RV also occurred, we assessed the levels of infectious RV from lysed alveolar macrophages by RV titration assay. A significant reduction of the presence of live infectious RV was observed at all time points in comparison with the amount of RV present following the 1 h infection period. Two days post infection, the level of live RV was almost zero (fig 1C).

Only infectious RV induces TNFα and IL8 release by human alveolar macrophages
RV-16 induced the release of TNFα from alveolar macrophages over 2, 5 and 10 days post infection. Exposure of alveolar macrophages to RV at an MOI of 1 significantly induced the release of TNFα at all three time points in comparison with non-infected control cells (fig 1D). When cells were infected with RV at an MOI of 0.1, the release of TNFα was significantly increased only on day 2 (fig 1D).
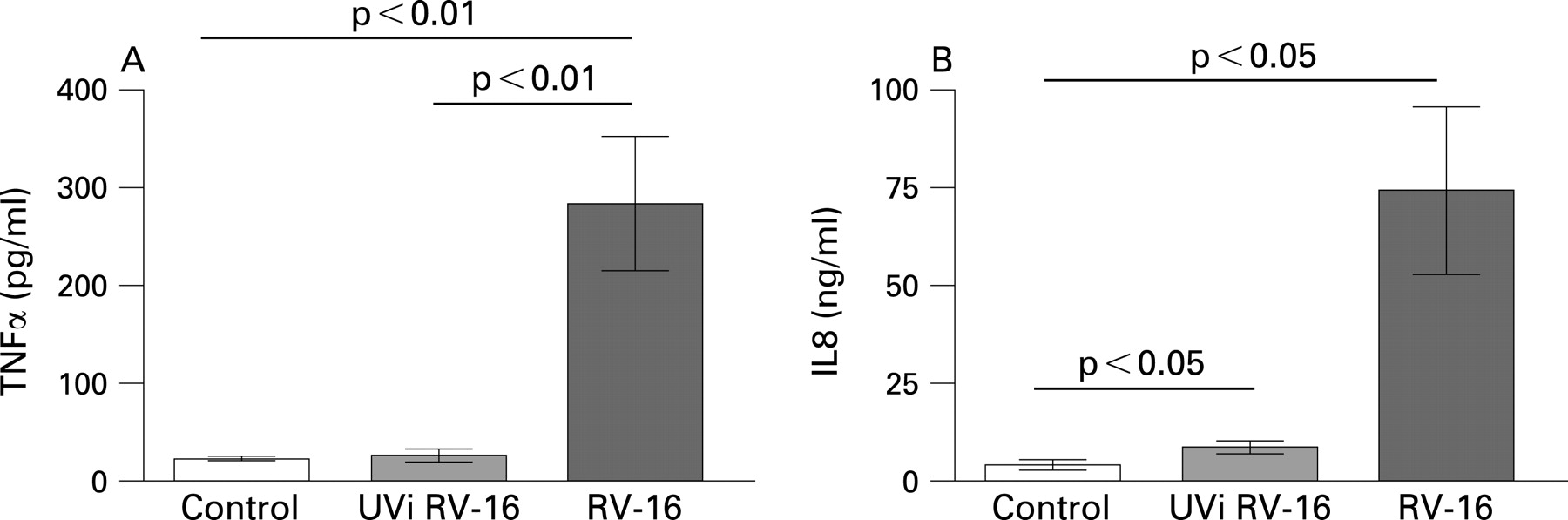
In our experiments, UVi-RV failed to induce TNFα and IL8 release from alveolar macrophages (fig 2A, B). Neither IFNγ nor eotaxin were present in the culture medium (data not shown). These data suggest that RV induction of TNFα and IL8 by alveolar macrophages is replication dependent.

RV downregulates alveolar macrophage immune responses to bacterial products
We then examined alveolar macrophage responses to gram negative and gram positive bacterial products in the presence and absence of prior RV infection. We examined TNFα and IL8 secretion (measured within the same tissue culture medium) from alveolar macrophages isolated from 16 subjects, exposed to RV-16 for 1 day and then further stimulated with LPS for 1 day. As depicted in fig 3A, LPS induced TNFα release. This LPS induced secretion was significantly inhibited when the cells had been pretreated with live infectious RV for 24 h (p<0.001). A similar inhibitory, and even more significant, effect of RV infection on LPS induced IL8 release by alveolar macrophages was observed for IL8, as shown in fig 3B (p<0.0001).
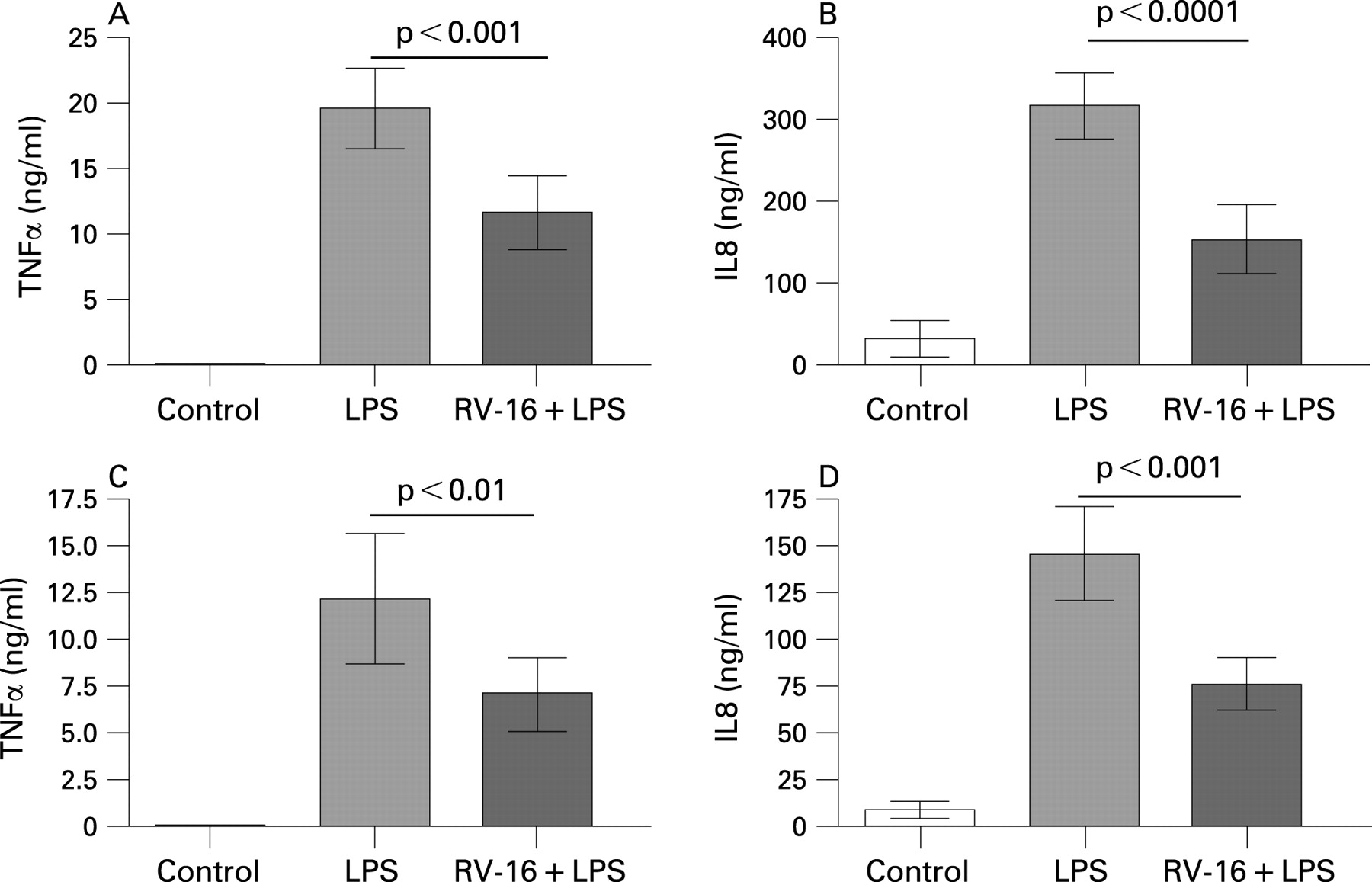
The experiments were repeated in alveolar macrophages which had been exposed to RV for 4 days, followed by LPS stimulation for 1 day. We observed that under this condition, LPS stimulated secretion of TNFα and IL8 by alveolar macrophages infected with RV was again significantly reduced in comparison with LPS alone (fig 3C, D), demonstrating that impairment of LPS responses persisted beyond the ability to detect live virus within macrophages. Interestingly, LPS stimulated cytokine release decreased with increased time in culture. We speculate that this is related to the in vitro age of the cells.
Effect of exposed UVi RV-16 on LPS induced cytokine release by alveolar macrophages
In contrast with infection with RV, pretreatment with UVi RV-16 (24 h) did not reduce LPS induced TNFα secretion (p>0.05, n = 8) or IL8 secretion (p>0.05, n = 8) (fig 4B), indicating that the impairment of LPS induced cytokine release by rhinovirus was likely replication dependent.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Minor group RV also downregulate LPS induced cytokine release by alveolar macrophages
A minor group RV, RV-2, was used (see online supplement) in a set of similar experiments. Pretreatment of alveolar macrophages with RV-2 for 24 h significantly inhibited LPS induced secretion of TNFα and IL8, in comparison with LPS stimulation alone, further supporting the observation that rhinovirus mediated inhibition of cytokine release was replication dependent.
RV inhibits LTA induced cytokine release by human alveolar macrophages
LTA stimulation (1 ng/ml) increased TNFα release (11.3 (10.6) ng/ml) by macrophages compared with control cells (0.02(0.002) ng/ml) but this was not statistically significant (p = 0.3297, n = 7). However, LTA significantly induced IL8 release (76.2 (19.2) ng/ml) by macrophages compared with controls (4.6 (0.96) ng/ml, p<0.05 n = 7) (see online supplement fig E1a).
While not statistically significant, LTA induced TNFα secretion by alveolar macrophages exposed to infectious RV-16 was reduced (from 11.3 (10.6) to 3.1 (2.5) ng/ml). In contrast, UVi RV-16 exposed alveolar macrophage (initial MOI of 1) caused a small but significant reduction in the secretion of IL8 in response to LTA, when compared with LTA alone (p<0.05, n = 7). However, exposure to infectious RV-16 demonstrated an even greater inhibition in LTA induced IL8 compared with UVi RV-16 exposed cells (p<0.01, n = 7) (see online supplement fig E1b).
Modulation of alveolar macrophage receptors for bacterial products by RV
To investigate the possibility that exposure to RV-16 downregulated the expression of cellular receptors for bacterial products (PRRs) on macrophages, this was assessed using flow cytometry. In untreated cells, expression of TLR2 (LTA receptor) but not CD14 or TLR4 (LPS receptors) was detectable by flow cytometry. Exposure to RV for 1 day did not downregulate cell surface TLR2 expression in comparison with non-infected cells (p>0.05, n = 5) (see online supplement fig E2.)
RV dependent secretion of anti-inflammatory mediators by alveolar macrophages
As cell surface TLR2 expression was not altered by RV infection, we next investigated whether RV impairment of antibacterial responses was mediated by RV induction of the anti-inflammatory mediators IL10 and prostaglandin E2 (PGE2). IL10 was not induced by RV exposure of alveolar macrophages (constitutive production 29.7 (5.1) pg/ml vs RV-16 MOI 1 26.4 (3.4) pg/ml, p = 0.18, n = 7). Based on preliminary data, we assessed the anti-inflammatory role of PGE238 on RV impaired cytokine secretion in macrophages, and the production of PGE2 was inhibited by the cyclooxygenase-2 inhibitors indomethacin (2.5 μM) and nimesulide (1.5 μM). As shown in the online supplement fig E3, cyclooxygenase-2 inhibitors did not reduce the RV dependant reduction in LPS induced cytokine release.
RV inhibits phagocytosis of E coli bioparticles but not latex beads in alveolar macrophages
An important innate immune response to bacteria by alveolar macrophages is phagocytosis. As RV exposure impaired the ability of macrophages to secrete TNFα and IL8 in response to bacterial products, we examined if RV exposure also downregulated phagocytosis by alveolar macrophages. As shown in the online supplement fig E4, RV reduced the phagocytosis of E coli bioparticles (p = 0.027, n = 3) but not latex beads (p>0.05, n = 5).
DISCUSSION
Infectious rhinovirus (RV) and RV RNA were detected in alveolar macrophages up to 3 and 10 days post exposure. We have shown that infectious RV but not UVi RV increased TNFα and IL8 release by alveolar macrophages and furthermore infectious RV reduced their ability to respond to LPS or LTA. This RV dependent impairment of the macrophage immune response was not mediated by autocrine production of the anti-inflammatory cytokines IL10 and PGE2, or by downregulation of the cell surface receptor for LTA. It is unlikely that RV infection induced a non-specific cellular downregulation as the phagocytic ability of the infected alveolar macrophages to ingest latex beads was not altered. However, since phagocytosis of E coli bioparticles was reduced, this further supports the notion that bacterial innate immune responses are reduced in alveolar macrophages infected with rhinovirus.
Since infectious RV virions could be isolated from infected macrophages up to 3 days after RV exposure, our data suggest that RV either actively infects human alveolar macrophages or is taken up via phagocytosis and is not immediately eliminated. Furthermore, RV RNA persisted in alveolar macrophages up to 10 days post RV exposure, indicating that RV survives and may even replicate at a low level for a limited time. These observations are in accordance with Gern et al, who reported that there was no increase in RV virions within 24 hours in alveolar macrophages.34 Thus it seems that the macrophage is able to allow but limit the replication of RV and perhaps act as a viral sink. If we assume that no RV replication occurs in the alveolar macrophage, and the observed decrease in infectious virions represents the natural RV decay, detection of RV RNA should mirror these events. Our findings suggest that RV is replicating in alveolar macrophages at a lower rate than that at which the virus is eliminated from the macrophage over this time period.
UVi-RV is incapable of replicating but it may bind and activate the host receptor in susceptible cells. Here we have shown that exposure of alveolar macrophages to UVi-RV failed to induce the production of TNFα and IL8, and therefore it can be assumed that their production was linked to ongoing RV replication. Others have shown replication independent responses in a variety of cells.39–42 A cell type specific response to RV infection at the MOI used is further supported by our observation that, using the same UVi-RV stocks and infection procedure, UVi-RV induced similar cytokine secretion to infectious RV in human airway smooth muscle cells.36 However, it is also possible that interaction between RV-16 and intercellular adhesion molecule 1 (ICAM-1) in alveolar macrophages may be altered following UV irradiation. Previous reports which have examined cytokine release from alveolar macrophages in response to exposure to UVi-RV have shown that both TNFα34 and MCP-1 (via P38 MAPK)43 were induced, perhaps suggesting that UV inactivation does not alter RV-ICAM interactions. In monocytes, IP-10 (via the JAK/STAT pathway) production has also been demonstrated following exposure to replication defective rhinovirus.44 The differences between these reports and our results could be accounted for by the 10-fold greater MOI of rhinovirus used in their studies.34 ,43 Further study will clearly be needed to clarify the importance of rhinovirus replication in alveolar macrophage function.
Cell death was not induced following viral exposure of alveolar macrophages, in agreement with other studies.34 In addition, the anti-inflammatory cytokine IL10 was not upregulated by RV infection of alveolar macrophages, in contrast with RV-14 exposure of blood monocytes in which significant upregulation of IL10 occurred.45
As an in vitro model of bacterial infection, we used LPS and LTA, components of the bacterial cell wall from gram negative and gram positive bacteria, respectively, to stimulate RV exposed and non-exposed alveolar macrophages. RV exposure resulted in a reduced ability to mount an immune response against bacterial products. This is a novel finding which provides a potential mechanism by which RV infection may foster bacterial superinfection. Even 4 days post RV exposure, the bacterial toxin induction of TNFα and IL8 was impaired. This indicates a long lasting effect of RV infection and might be related to the sustained presence of RV RNA in the host cell. Using UVi-RV, only LTA induced IL8, but not TNFα, secretion was inhibited compared with toxin stimulation alone. However, considerably greater impairment of LTA dependent IL8 release was observed when cells were exposed to infectious RV. This suggests that LTA induced IL8 production is partly ICAM-1 dependent, but largely replication dependent.
We were unable to detect cell surface expression of either CD14 or TRL4. This is most likely the result of the staining technique we used, as CD14 expression on alveolar macrophages is undetectable unless autofluorescence is blocked.46 We chose not to block autofluorescence, as this procedure would have permeabilised the cell membrane thereby introducing the possibility that intracellular and extracellular CD14 would be detected. Other studies indicate that CD14 cell surface expression on alveolar macrophages (5–50%) is considerably lower than that found on blood monocytes.47 ,48 However, we were able to detect the cell surface expression of TLR 2, the cellular receptor for LTA,49 ,50 and found no downregulation following RV exposure. We concluded that bacterial toxin receptor downregulation by RV is unlikely to be the cause of the observed impairment of cytokine release.
We further assessed if the effect of RV infection on alveolar macrophages reflected a general downregulation of the function of the host cells using phagocytosis as an indicator. RV exposure did not modulate the phagocytic ability of alveolar macrophages to ingest latex beads compared with non-RV exposed macrophages, but phagocytosis of bacterial particles was significantly impaired. This supports our hypothesis that RV infection of alveolar macrophages would promote the occurrence of a bacterial superinfection.
As exposure to UVi RV did not alter the LPS induced secretion of TNFα and IL8 by alveolar macrophages, we can assume that the RV dependent TNFα and IL8 downregulation observed was not mediated through ICAM-1. To confirm this, we exposed alveolar macrophages to a minor group RV (which does not infect cells via ICAM-1), RV-2, and found that LPS induced TNFα and IL8 secretion was reduced, as was found with RV-16.
In summary, this study provides evidence for a novel unidentified mechanism by which RV can impair the innate immune response in alveolar macrophages and may thereby provide an environment that facilitates additional bacterial infection.
Acknowledgments
We acknowledge the collaborative effort of the cardiopulmonary transplant team and the pathologists at St Vincent’s Hospital, Sydney and the thoracic physicians and pathologists at Royal Prince Alfred Hospital, Concord Repatriation General Hospital and Strathfield Private Hospital and Rhodes Pathology, Sydney. We also acknowledge the contribution of Joanne Thompson for excellent technical assistance.
REFERENCES
Supplementary materials
web only appendices 63/6/519
Files in this Data Supplement:
Footnotes
Funding: This work was supported by the National Health and Medical Research Council of Australia (NH&MRC).
Competing interests: None.
Ethics approval: Ethical approval was obtained.