Article Text

Abstract
Exacerbations of chronic obstructive pulmonary disease (COPD) are associated with increased morbidity and mortality. The effective management of COPD exacerbations awaits a better understanding of the underlying pathophysiological mechanisms that shape its clinical expression. The clinical presentation of exacerbations of COPD is highly variable and ranges from episodic symptomatic deterioration that is poorly responsive to usual treatment, to devastating life threatening events. This underscores the heterogeneous physiological mechanisms of this complex disease, as well as the variation in response to the provoking stimulus. The derangements in ventilatory mechanics, muscle function, and gas exchange that characterise severe COPD exacerbations with respiratory failure are now well understood. Critical expiratory flow limitation and the consequent dynamic lung hyperinflation appear to be the proximate deleterious events. Similar basic mechanisms probably explain the clinical manifestations of less severe exacerbations of COPD, but this needs further scientific validation. In this review we summarise what we have learned about the natural history of COPD exacerbations from clinical studies that have incorporated physiological measurements. We discuss the pathophysiology of clinically stable COPD and examine the impact of acutely increased expiratory flow limitation on the compromised respiratory system. Finally, we review the chain of physiological events that leads to acute ventilatory insufficiency in severe exacerbations.
- COPD, chronic obstructive pulmonary disease
- DH, dynamic hyperinflation
- EELV, end expiratory lung volume
- EFL, expiratory flow limitation
- FEV1, forced expiratory volume in 1 second
- FRC, functional residual capacity
- FVC, forced vital capacity
- IC, inspiratory capacity
- PEFR, peak expiratory flow rate
- RV, residual volume
- TLC, total lung capacity
- Ve, minute ventilation
- Vt, tidal volume
- chronic obstructive pulmonary disease
- pathophysiology
- exacerbations
Statistics from Altmetric.com
- COPD, chronic obstructive pulmonary disease
- DH, dynamic hyperinflation
- EELV, end expiratory lung volume
- EFL, expiratory flow limitation
- FEV1, forced expiratory volume in 1 second
- FRC, functional residual capacity
- FVC, forced vital capacity
- IC, inspiratory capacity
- PEFR, peak expiratory flow rate
- RV, residual volume
- TLC, total lung capacity
- Ve, minute ventilation
- Vt, tidal volume
The progression of chronic obstructive pulmonary disease (COPD) is associated with increasing frequency and severity of exacerbations. COPD exacerbations are clearly linked to impoverished health status and can be life threatening, particularly in patients with advanced disease.1 In some patients exacerbations result in prolonged activity limitation and can quickly reverse the hard won benefits of exercise training programmes. The clinical diagnosis of COPD exacerbations is currently made on the basis of sustained worsening of the common respiratory symptoms. For the purpose of this review, we will use the simple pragmatic definition of a COPD exacerbation recently provided by the Canadian Thoracic Society: “a sustained worsening of dyspnea, cough or sputum production leading to an increase in the use of maintenance medications and/or supplementation with additional medications”.2 The clinical manifestations of COPD exacerbations are highly variable and reflect broad heterogeneity in the underlying pathophysiology of COPD as well as diversity in the nature and effect of the inciting agent. Expiratory flow limitation (EFL), as a consequence of airway inflammation, is the pathophysiological hallmark of COPD. Exacerbations fundamentally reflect acute worsening of EFL, and there is evidence for both increased airway inflammatory activity and worsening airway obstruction as plausible explanations.3
It is reasonable to assume that worsening airway inflammation is the primary inciting event of COPD exacerbations and may be caused by bacteria, viruses, or environmental pollutants, including cigarette smoke. The mechanisms of acute worsening of EFL in the absence of airway inflammation, however, are less well understood. Temporal day to day, within patient variability in the extent of EFL may be considerable, even in patients with clinically stable disease. Daily bronchodilator requirements are often variable and airway obstruction may become refractory to the usual bronchodilator treatment for periods of time. The recent evidence that long acting bronchodilators and mucolytic agents are associated with reduced frequency and severity of exacerbations bolster the notion that factors other than airway inflammation may be important.4,5 Newer bronchodilators, by achieving sustained airway patency and lung volume reduction, are associated with better dyspnoea control and fewer episodes of symptomatic deterioration requiring short acting “reliever” bronchodilators. The mechanisms of protection from symptomatic exacerbations are not known, but improved sputum clearance, reduced variability in airway smooth muscle tone, and altered (increased) dyspnoea thresholds as a result of sustained lung deflation are possible explanations.
Our knowledge of the pathophysiological basis of COPD exacerbations continues to grow. Detailed mechanical studies in hospitalised patients with acute respiratory failure undergoing mechanical ventilation have provided a clearer understanding of the pathophysiology of severe exacerbations.6 In this situation, the main mechanism is critical EFL with attendant lung hyperinflation, which leads to serious mechanical consequences. Considerably less is known about the mechanisms underlying symptomatic deterioration during mild to moderate exacerbations which are much more commonly encountered in clinical practice. However, it is reasonable to assume that moderate exacerbations share the same basic physiological underpinnings as severe exacerbations, and that the difference is mainly one of degree.
PULMONARY FUNCTION
The routine use of pulmonary function tests to aid in the diagnosis and characterisation of severity of stable COPD has been suggested by recent clinical practice guidelines.2 However, the usefulness of spirometric or pulmonary function tests for the diagnosis of acute exacerbations or for the prediction of clinical outcomes remains unknown. Unlike asthma, where indices of expiratory flow limitation (such as peak expiratory flow rate (PEFR) or forced expiratory volume in 1 second (FEV1)) have a well defined role in patient management, the changes in spirometric variables observed during exacerbations of COPD are variable,7,8,9,10,11,12 and the time course of recovery of such physiological parameters following an exacerbation does not always follow that of symptom recovery.3 Lastly, given the heterogeneity of the structural abnormalities and pathophysiology which characterise COPD, the overall degree of impairment during exacerbations may not be adequately reflected in simple spirometric parameters.
Time course of change of spirometric parameters during COPD exacerbations
Seemungal and colleagues prospectively followed a cohort of 101 patients with moderate to severe COPD (mean FEV1 41.9% predicted) for a period of 2.5 years.13 All subjects recorded daily symptoms and PEFR on diary cards, and a smaller subset (n = 34) also recorded daily spirometry. During the follow up period a total of 504 exacerbations were diagnosed based on symptomatic deterioration. Before the exacerbation there were no significant changes in either PEFR or FEV1, suggesting that these variables are not useful in predicting the onset of an exacerbation, at least not in the ambulatory patient population. Furthermore, at the onset of the exacerbation, the magnitude of change in PEFR, FEV1, and forced vital capacity (FVC) were small (8.6 l/min, 24.0 ml, and 76.0 ml, respectively; fig 1). There was a weak but statistically significant correlation between greater falls in PEFR at onset of the exacerbation and symptoms of increased dyspnoea (r = 0.12, p = 0.014). Stronger associations were found between greater decreases in PEFR (r = −0.56), FEV1 (r = −0.56), and FVC (r = −0.50) and longer time to recovery of these variables (p<0.001). After exacerbation the median time to recovery of PEFR (6 days) was similar to that of symptomatic recovery (7 days). At 35 days after the exacerbation, however, the PEFR had returned to baseline in only 75.2% of exacerbations and approximately 14% of exacerbators had incomplete recovery of symptoms. Even at 91 days after the exacerbation 7.1% of exacerbations had not recovered PEFR to pre-exacerbation levels.

Time course of recovery of peak expiratory flow rate (PEFR) after exacerbation. A cohort of 101 patients with moderate to severe COPD (mean FEV1 41.9% predicted was followed prospectively until the time of exacerbation (day 0) and then during recovery. Daily median PEFR is plotted against time and expressed as a percentage of baseline (pre-exacerbation) levels. A significant proportion of subjects did not attain their baseline value of PEFR, even 30 days after the onset of exacerbation. Reproduced with permission from Seemungal et al.13
Other studies that have followed patients prospectively after presentation with an exacerbation have found similarly modest changes in PEFR and FEV1 during recovery,7,8,9,10,12 although patients who have sufficiently severe exacerbations to require hospitalisation may show greater changes in FEV1 during convalescence.11,14 The recovery of FEV1 after COPD exacerbations is also influenced by therapeutic interventions. A recent meta-analysis of the use of systemic corticosteroids in the treatment of acute exacerbations found that, within the first 72 hours of treatment, FEV1 was significantly higher in patients who received systemic corticosteroids than in those given placebo (weighted mean difference 140 ml, 95% CI 80 to 200, p = 0.00002).15
Spirometric measures as predictors of clinical outcome
A prospective study involving 340 patients with severe COPD (mean FEV1 36% predicted at time of exacerbation) found a relationship between higher values of FEV1 and a reduction in the need for readmission following an exacerbation (hazard ratio (HR) 0.97 per percentual unit of FEV1 % predicted, 95% CI 0.96 to 0.99; p<0.001),16 which supported similar results obtained in an earlier case-control analysis.17 Niewoehner and colleagues also found that a higher FEV1 at exacerbation onset was associated with a lower risk of treatment failure at 30 days (defined as death, intubation, readmission for COPD, or intensification of drug treatment (odds ratio (OR) 0.87 for a 100 ml increase in FEV1, 95% CI 0.79 to 0.96).8 In addition, the change in FEV1 (ΔFEV1) between study entry and day 2 of treatment was also predictive of treatment success, subjects with an increase in FEV1 by day 2 of >100 ml being less likely to experience treatment failure (OR 0.8, 95% CI 0.69 to 0.92). Collectively, subjects with an entry FEV1 and a day 2 ΔFEV1 that were both above the median (>750 ml and >100 ml increase, respectively) had a significantly lower rate of treatment failure than those in whom both values fell below the median (8% v 45%, p<0001).8 In a prospective study, larger changes in PEFR, FEV1, and FVC occurring at the time of the exacerbation were also significantly related to the length of recovery time for these parameters, with larger changes associated with longer times to physiological recovery.13 However, many of these studies are limited by the lack of pre-exacerbation baseline values of either PEFR or FEV1. Thus, the increased incidence of adverse outcomes in patients presenting with lower values of FEV1 at the time of exacerbation14 may reflect the intuitive notion that patients with more severe disease at baseline (and therefore lower FEV1 at baseline) do worse during an exacerbation than patients with a relatively higher FEV1.
Spirometric data to predict the need for mechanical ventilation
In a retrospective study Vitacca et al evaluated the ability of spirometric data to predict the need for mechanical ventilation in patients presenting with an exacerbation of COPD.18 Using discriminant analyses they found that the FEV1/FVC ratio, as well as vital capacity (% predicted) or FVC (% predicted), could discriminate between patients who required mechanical ventilation and those who did not. Other physiological variables including tidal volume (Vt), breathing frequency, and minute ventilation (Ve) did not differ significantly between the two groups, and did not predict the need for mechanical ventilatory support.
PATHOPHYSIOLOGY OF COPD EXACERBATIONS
EFL is a pathophysiological hallmark of COPD. Patients with COPD are said to be flow limited when the expiratory flow they generate during tidal respiration represents the maximal possible flows that they can generate at that volume. In flow limited patients the time available for lung emptying (expiratory time) during spontaneous breathing is often insufficient to allow end expiratory lung volume (EELV) to decline to its natural relaxation volume leading to lung overinflation.19 Thus, in flow limited patients, EELV becomes dynamically rather than statically determined, and essentially becomes a continuous variable that fluctuates widely depending on the extent of EFL and the prevailing ventilatory demand. Dynamic hyperinflation (DH) refers to acute and variable increase in EELV above its baseline value. DH occurs during exercise in flow limited patients as inspired tidal volume increases and expiratory time decreases, and is associated with severe mechanical constraints on ventilation and perceived respiratory discomfort.19 DH also occurs during acute bronchoconstriction in asthma. In this setting, the reduction in inspiratory capacity (IC), which reflects the increase in EELV, correlates strongly with the perception of inspiratory difficulty.20,21
During COPD exacerbations airways resistance is abruptly increased (due to bronchospasm, mucosal oedema, and sputum inspissation) and this worsens EFL. The time constant for lung emptying (given by the product of resistance and compliance) is therefore prolonged and EELV is dynamically increased. Furthermore, during an exacerbation, patients tend to adopt a rapid shallow breathing pattern which further limits the time available for lung emptying, thus promoting greater DH in a vicious cycle. In fact, any acute increase in ventilation (such as occurs with anxiety or transient hypoxaemia) can be associated with DH in flow limited patients. There is abundant evidence that acute DH may be life threatening during severe exacerbations of asthma or COPD.22 A summary of the deleterious effects of DH is shown in fig 2.

Schematic of the negative consequences of dynamic hyperinflation. During exacerbation, dynamic hyperinflation develops as a consequence of worsening expiratory flow limitation. Abbreviations are all defined in the text.
Negative mechanical effects of dynamic hyperinflation
In contrast to the situation in health where operating lung volumes remain positioned on the favourable steep portion of the respiratory system compliance curve, tidal breathing during an exacerbation in a patient with COPD may be shifted upwards closer to total lung capacity (TLC) as a consequence of DH (fig 3). Although this optimises expiratory flows, it has the deleterious effect of forcing the respiratory system to operate on the flatter part of the compliance curve where progressive pressure increases generate smaller incremental volume changes. In effect, severe DH imposes a “restrictive” mechanical constraint on the respiratory system, where tidal volume expansion is limited in the face of increasing inspiratory effort.

Schematic of mechanical effects of COPD exacerbation. Representative pressure-volume plots during (A) stable COPD and (B) COPD exacerbation. During exacerbation, worsening expiratory flow limitation results in dynamic hyperinflation with increased end expiratory lung volume (EELV) and residual volume (RV). Corresponding reductions occur in inspiratory capacity (IC) and inspiratory reserve volume (IRV). Total lung capacity (TLC) is unchanged. As a result, tidal breathing becomes shifted rightward on the pressure-volume curve, closer to TLC. Mechanically, increased pressures must be generated to maintain tidal volume (Vt). At EELV during exacerbation, intrapulmonary pressures do not return to zero, representing the development of intrinsic positive end expiratory pressure (PEEPi) which imposes increased inspiratory threshold loading (ITL) on the inspiratory muscles (inset); during the subsequent respiratory cycle, PEEPi must first be overcome in order to generate inspiratory flow.
While the ventilatory muscles partially adapt to chronic hyperinflation to preserve their force generating capacity during resting breathing,23–25 these adaptations can become quickly overwhelmed in the setting of suddenly increased DH.19,26–29 During COPD exacerbations the respiratory muscles which are already burdened by increased resistive loading become subject to increased elastic loading—that is, greater effort required for a given change in volume. Acute DH further shortens the inspiratory muscles, particularly the diaphragm, and causes functional muscle weakness. Exposure to oxidative stress and local activation of proteases may also result in diaphragmatic injury during periods of increased inspiratory loading and result in inspiratory muscle dysfunction.30 As a consequence of EFL, intrapulmonary pressures are positive at the end of expiration representing intrinsic or auto-positive end expiratory pressure (PEEPi). The presence of PEEPi means that the inspiratory muscles must first overcome the combined inward recoil of the lung and chest wall before inspiratory flow can be initiated. Thus, PEEPi essentially acts as an inspiratory threshold load and has been measured to be as much as 6–9 cm H2O during quiet breathing at rest in clinically stable but hyperinflated COPD patients.31,32 During acute-on-chronic pulmonary hyperinflation of COPD exacerbations, the attendant tachypnoea also results in reduced dynamic lung compliance (Cdyn) reflecting the effect of the marked time constant inequalities of alveolar units throughout the lungs in this disease.33
The net effect of this increased loading and functional weakness of the inspiratory muscles is that the effort required for tidal inspiration represents a relatively high fraction of the maximal possible effort that the patient can develop at that lung volume. This increased effort may be directly perceived (via central corollary discharge) as unpleasant if it exceeds a certain threshold.34 The neural drive to breathe is usually preserved in COPD (even in many patients with chronic compensated hypercapnic acidosis) and actually increases during times of physiological stress such as during exercise or exacerbations.35,36 Central drive increases further in the presence of critical arterial oxygen desaturation, carbon dioxide retention, or acidosis, and is also stimulated by fever or increased sympathetic nervous system activation. However, during COPD exacerbations the mechanical output of the flow limited respiratory system may not increase in proportion to neural drive, resulting in neuromechanical dissociation or uncoupling of the respiratory system. This disparity between central drive and the mechanical response as a consequence of acute DH has been implicated in the genesis of dyspnoea (or its dominant qualitative dimensions) in COPD patients during exercise,37,38 and in asthmatics during acute bronchoconstriction.20 It is reasonable to assume that similar mechanisms explain dyspnoea during COPD exacerbations.
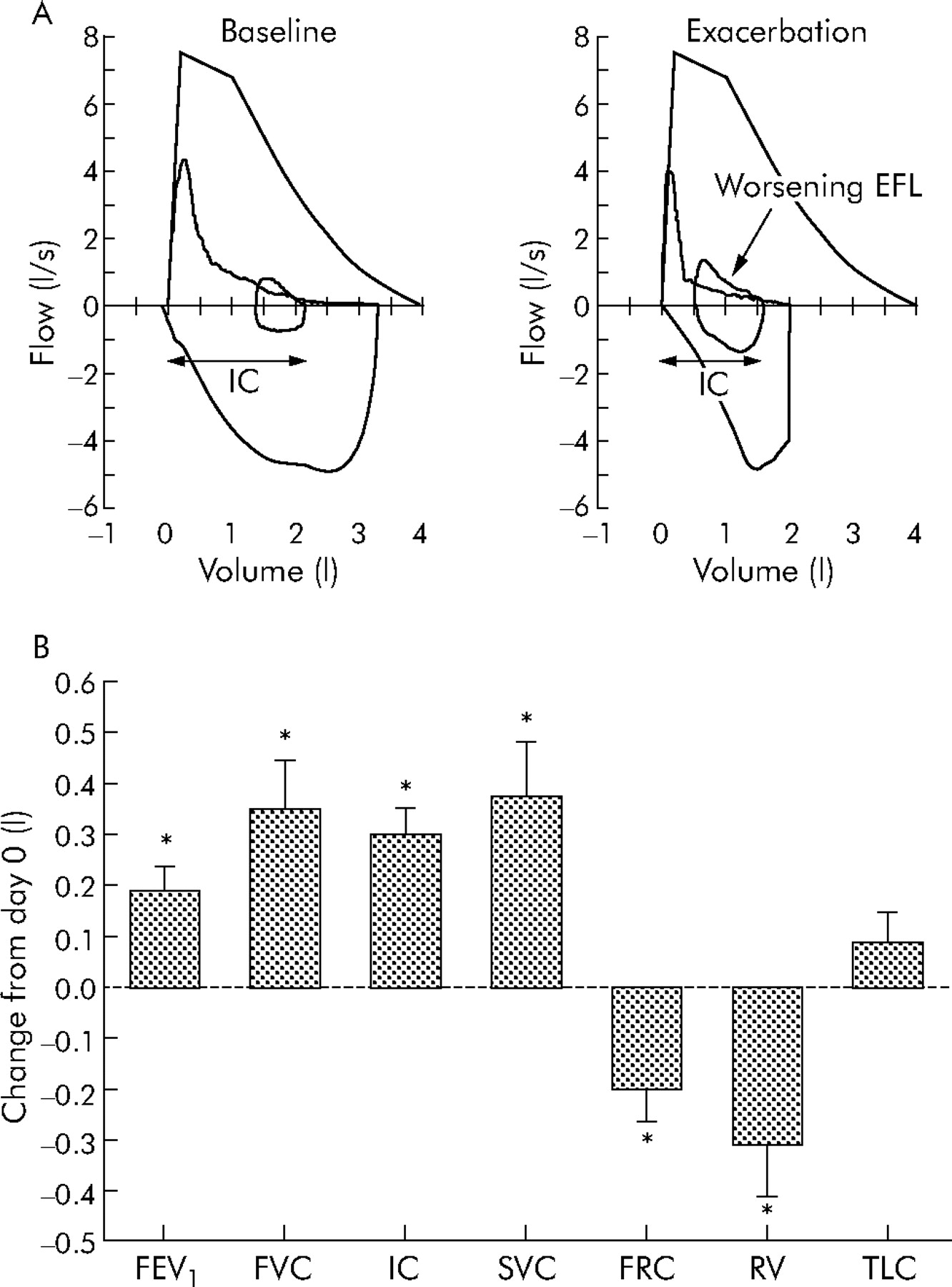
While there is extensive evidence for the deleterious clinical consequences of DH in patients presenting with acute respiratory failure as a result of COPD exacerbations, there is very little information on the existence or clinical impact of DH in mild to moderate exacerbations. A recent study from our laboratory evaluated the changes occurring in various physiological parameters during moderate exacerbations not associated with ventilatory failure.7 Twenty patients with moderately severe COPD were studied within 72 hours of initial worsening of symptoms (primarily dyspnoea) consistent with an exacerbation, and underwent full pulmonary function testing and symptom assessment using the dyspnoea domain of the Chronic Respiratory Disease Questionnaire (CRQ-dyspnoea). At the time of study entry (day 0) subjects were “very” short of breath (CRQ-dyspnoea 2.4 (0.3)) and had significant airflow obstruction (FEV1 41(3)% predicted) and hyperinflation (FRC 164 (7)% predicted). At the end of the follow up period most patients reported that their dyspnoea had returned to the baseline (pre-exacerbation) level, and overall there was significant and clinically relevant improvement in the mean CRQ-dyspnoea score (by 1.6 (0.4) units). In conjunction with the improvement in symptoms, indices of hyperinflation and gas trapping were also improved (fig 4) with significant reductions in FRC (by 200 (60) ml) and RV (by 310 (100) ml). Inspiratory capacity (IC) also improved significantly during symptomatic recovery with an increase of 300 (50) ml noted between day 0 and the final follow up visit. Importantly, TLC did not change during recovery, which suggests that the IC (which can be obtained from simple spirometric measurements) can be used to reliably detect changes in EELV during COPD exacerbations. Although significant improvements were also noted in expiratory flows and volumes during recovery (PEFR improved by 0.6 (0.14) l/s and FEV1 improved by 190 (50) ml), there was no significant change in the FEV1/FVC ratio. This may reflect a proportional change in both FEV1 and FVC during recovery, but it may also be explained by improvements in expiratory flow rates occurring as a consequence of improved expiratory volumes due to a reduction in the mechanical constraints imposed by lung hyperinflation. Breathing pattern analysis of subjects in this study did not show any significant differences in breathing frequency or expiratory time during recovery, which suggests that the propensity to develop DH during COPD exacerbations may be reflective of worsening EFL rather than a shortening of the expiratory duty cycle per se.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Magnitude of change in lung function parameters during recovery from exacerbation. (A) Representative flow-volume loops from a patient obtained at baseline (that is, before the exacerbation) and after onset of symptoms compatible with exacerbation. During exacerbation there is evidence of worsening expiratory flow limitation (EFL, arrow) resulting in hyperinflation with an increased end expiratory lung volume (EELV) and reduced inspiratory capacity (IC). (B) Change in lung function parameters during recovery from moderate exacerbations in 20 patients. Subjects were studied (day 0) within 72 hours of symptomatic deterioration. Data shown are change from initial (day 0) assessment. FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; IC, inspiratory capacity; SVC, slow vital capacity; FRC, functional residual capacity; RV, residual volume; TLC, total lung capacity. Modified with permission from Parker et al.7
In another recent study, Stevenson et al12 evaluated changes in spirometric parameters during recovery in 22 patients with COPD exacerbations required admission to hospital and found similar results. Compared with values obtained at the time of admission, the mean post-bronchodilator FEV1 improved by 0.20 l and the mean IC improved by 0.42 l after 42 days of follow up. Furthermore, there was no change in the FEV1/FVC ratio during the follow up period and, as in the study by Parker et al,7 the authors concluded that favourable changes in volume during recovery resulted in improved expiratory flows.
The magnitude of change in IC (approximately 20% of baseline value) found in these studies may have important clinical consequences. There is evidence that in flow limited patients with reduced resting IC (<80% predicted), further acute reductions in IC as a result of DH lead to a limited ability to increase Vt and ventilation when demand suddenly increases as, for example, in exercise.39,40 The corollary of this is that increases in resting IC (between 200 and 400 ml) after bronchodilator treatment were found to correlate strongly with improvements in exertional dyspnoea and exercise endurance in patients with moderate to severe COPD.40–42
ACUTE RESPIRATORY FAILURE DURING COPD EXACERBATIONS
Although patients with severe COPD may be able to maintain acceptable indices of gas exchange during times of stability, acute decompensation in the setting of an exacerbation is relatively common. The pathophysiological adaptive mechanisms that characterise stable disease can be quickly overcome, and the ability of the patient to further adapt to the added physiological stress that accompanies an exacerbation is influenced by the degree of physiological dysfunction during the stable state. Patients with more severe or advanced disease therefore have less physiological reserve and are thus prone to develop acute respiratory failure, even in the setting of a relatively mild precipitant such as a common cold. Alternatively, patients with milder disease may still experience respiratory failure if the precipitating illness is sufficiently severe.
Acute respiratory failure in the setting of a COPD exacerbation is characterised by worsening hypoxaemia and often presents with variable degrees of carbon dioxide retention and acidaemia. Hypoxaemia probably results from worsening ventilation-perfusion (V/Q) mismatching, often with modest increases in shunt fraction.22 However, even in severe exacerbations, hypoxaemia is usually easily corrected by modest doses of supplemental oxygen. Barbera et al43 evaluated the mechanisms underlying abnormal gas exchange in 13 patients with severe COPD (mean FEV1 29% predicted) during an acute exacerbation. At the time of initial assessment, these patients were severely hypoxaemic (mean arterial oxygen tension (Pao2) 44 mm Hg (5.9 kPa)), hypercarbic (mean arterial carbon dioxide tension (Paco2) 55 mm Hg (7.3 kPa)), and mildly acidaemic (arterial pH 7.35), but did not require mechanical ventilation. Using the multiple inert gas elimination technique (MIGET), these authors found that worsening of V/Q relationships with increased V/Q mismatching accounted for nearly 50% of the observed hypoxaemia. These effects were further compounded by a reduction in the mixed venous oxygen tension (Mvo2), presumably reflecting increased oxygen consumption (V˙o2) due to increased work of the respiratory muscles but, in these patients, the impact of the reduced Mvo2 on arterial oxygenation was partially attenuated by an increase in cardiac output. Ve was also slightly increased during exacerbation, which was largely attributable to an increase in breathing frequency, and the authors therefore concluded that the observed hypercarbia in these patients was also a consequence of worsening V/Q mismatching rather than alveolar hypoventilation.43
The presence of decompensated hypercarbia during an acute exacerbation is an important prognostic consideration and correlates with the risk of both short and long term mortality.44,45 In some patients the use of higher amounts of supplemental oxygen may result in worsened CO2 retention. Although the mechanisms of this are debated,46 there is evidence to support both worsening of V/Q mismatching (as a result of the loss of hypoxic vasoconstriction)47,48 as well as a reduced central drive to breathe (as a result of the loss of hypoxaemic ventilatory drive).49 The propensity to develop hypercarbia is also influenced by relative increases in dead space50 which results in a relative alveolar hypoventilation despite increases in respiratory drive. In advanced COPD, physiological dead space (wasted ventilation) is increased as a consequence of underlying V/Q mismatch. As a result, patients with COPD must adopt a higher minute ventilation in order to keep alveolar ventilation (and hence Paco2) constant. Although these adaptive mechanisms may serve to maintain Paco2 within normal limits during times of relative disease stability, they may not be adequate to maintain gas exchange homeostasis in the face of increased physiological stress such as during an exacerbation or exercise. In the setting of EFL, DH and subsequent constraint of tidal volume expansion would be expected to further limit the ability of the respiratory system to adequately remove carbon dioxide from the blood, and this situation is made worse by an increase in V˙co2 as a result of increased metabolic demands.
The development of a rapid shallow breathing pattern during an exacerbation or exercise probably reflects the presence of restrictive mechanics and increased elastic loading.19 A study of 20 patients with advanced COPD (mean FEV1 34% predicted) by O’Donnell et al48 found that the tendency to develop carbon dioxide retention during exercise could be predicted by the degree of DH. Specifically, the subset of patients who retained carbon dioxide during the increased metabolic load of exercise showed earlier attainment of peak alveolar ventilation and greater DH relative to the non-retaining cohort, and there was good correlation between the EELV/TLC (as an index of dynamic hyperinflation) and Paco2 (r = 0.562, p<0.0005). The authors concluded that carbon dioxide retention occurred, at least in part, as a result of greater dynamic mechanical constraints in the presence of a fixed high physiological dead space. It is tempting to speculate that patients undergoing COPD exacerbation—who may display worsening of underlying EFL and have a similar rapid shallow breathing pattern—would also be prone to developing carbon dioxide retention as a result of mechanical limitation.
The pathophysiology of severe COPD exacerbations requiring mechanical ventilation is now well understood. Increased airways resistance, usually as a result of worsening airway inflammation, results in critical EFL and DH with dramatically increased loading and functional weakness of the inspiratory muscles. The accessory muscles of breathing are maximally recruited and significant thoracoabdominal dysynchrony is often evident. Inspiratory threshold loads increase substantially (values between 13 and 20 cm H2O have been recorded) and this has been suggested to account for nearly 60% of the increased static inspiratory work of breathing during an exacerbation.51–54 Dynamic lung compliance becomes precipitously reduced as a result of the accompanying tachypnoea (and reduced inspiratory time) and, in conjunction with the increased airways resistance, also contributes importantly to the increased dynamic inspiratory work.6,51–54
The role of respiratory muscle fatigue as a possible contributor to acute respiratory failure in the setting of a COPD exacerbation is the subject of debate. Fatigue—defined as a decrease in generated muscle force in response to a given neural stimulus—might be expected in the situation of an acute exacerbation, where additional resistive, elastic, and inspiratory threshold loads are imposed on the weakened inspiratory muscles. Conversely, however, it has been postulated that, in patients with chronic hyperinflation, the endurance of the inspiratory muscles may actually be increased,27,55 and there is evidence to suggest that diaphragmatic fatigue does not occur even during exhaustive exercise.56,57 However, in severe COPD exacerbations associated with prolonged near maximal loading of the inspiratory muscles and alterations in the metabolic milieu (acute hypoxia, hypercapnia, and acidosis combined with compromised blood flow), fatigue is almost inevitable.
Mechanical ventilation may be required for severe exacerbations presenting with acute respiratory failure. There is now ample evidence58–60 that non-invasive positive pressure ventilation (NPPV), when applied appropriately, reduces the need for endotracheal intubation, confers a clear mortality benefit, and results in significant improvements in dyspnoea and physiological variables in the setting of an acute exacerbation. NPPV improves gas exchange during COPD exacerbations, with an increase in Pao2, a decrease in Pa–ao2, and a reduction in Paco2.61,62 These effects are largely mediated by increases in alveolar ventilation as a result of increased minute ventilation, which in turn occurs due to increased tidal volumes; there appears to be little change in V/Q relationships as a result of NPPV when used in this setting.62 NPPV also offloads the inspiratory muscles during an exacerbation and allows significant reductions in work of breathing.63,64 This occurs partly as a consequence of the applied (or extrinsic) PEEP which counterbalances the effects of PEEPi, thereby reducing the inspiratory threshold load.63 Patient-ventilator asynchrony is also reduced since inspiratory efforts are more likely to initiate a supported breath from flow triggered ventilators.65
Cardiac dysfunction during severe COPD exacerbations
In flow limited patients with COPD, the acute development of DH (and PEEPi) may reduce right ventricular (RV) preload as a result of impaired venous return.66 Furthermore, pulmonary artery pressures are generally higher at any cardiac output in patients with COPD than in healthy subjects. This probably occurs as a consequence of emphysematous destruction of vascular beds within the lungs, aggravated by pulmonary arterial hypoxaemia due to relative alveolar hypoventilation or V/Q mismatching, and contributes to increased right ventricular afterload.67–71 In patients with acute DH there may be a further increase in pulmonary vascular resistance associated with breathing at lung volumes close to TLC.67,68,71,72 This has been shown to occur during exercise and a similar phenomenon is likely to occur during COPD exacerbations.
Left ventricular (LV) diastolic function may also be adversely affected by the increases in right ventricular end diastolic pressures as a consequence of ventricular interdependence.55,73 In this case, the interventricular septum is shifted towards the left ventricle during diastole to accommodate the increased end diastolic right ventricular volume which may, in turn, impair left ventricular filling.72,74 In contrast to diastolic function, left ventricular systolic function is often preserved in COPD in the absence of co-morbidities such as ischaemic heart disease.72,74 However, in the setting of the acute and progressive increases in negative intrathoracic pressure which occur during COPD exacerbations (as a result of combined resistive and elastic loading), left ventricular afterload would be expected to be increased as a consequence of the larger imposed transmural pressure gradient. At the upper extremes of the respiratory compliance curve, these pressures may be substantial and significantly increase the work of the left ventricle to maintain cardiac output.
CONCLUSIONS
The prompt recognition and treatment of acute exacerbations are recognized in all recent practice guidelines as an important goal in the management of COPD. Our understanding of the cascade of physiological events that culminate in acute life threatening respiratory failure during COPD exacerbations continues to improve. Critical expiratory flow limitation with consequent severe dynamic lung hyperinflation appears to be the pivotal abnormality and this has disastrous implications with respect to the mechanical and gas exchange functions of the respiratory system. Associated deleterious cardiopulmonary interactions are also undoubtedly important in some patients. Much less is known about the aetiology and pathophysiology of less severe exacerbations but, in all likelihood, the basic underlying mechanisms are similar although the degree of neuromechanical dissociation is less marked. Based on previous work on the mechanisms of dyspnoea in asthma and in COPD during exercise, it is tempting to speculate that this dominant symptom of moderate COPD exacerbations also has its neurophysiological origin in the disparity between central drive and the “restricted” mechanical response of the flow limited respiratory system.
Currently we rely primarily on the report by patients of worsening symptoms, together with physical examination, to diagnose and evaluate the severity of COPD exacerbations. This assessment is generally not confirmed by any objective measure of physiological impairment although, in a severe exacerbation, arterial blood gas tensions may be used to guide management. Traditional tests of forced maximal expiratory flow rates have not been shown to predict exacerbations reliably or to grade their severity. The development of a simple and accurate diagnostic test that can measure physiological deterioration and predict clinical outcome remains an elusive goal. There is preliminary information that IC measurements—which indirectly measure the extent of lung hyperinflation during COPD exacerbations—may provide additional and clinically relevant information.
REFERENCES
Footnotes
-
Funding: none.
-
Competing interests: none declared.