Article Text

Abstract
Background: The effect of hypoxia on the formation of superoxide (O2−), the expression of gp91phox and endothelial NO synthase (eNOS) were studied in pig intact pulmonary artery (PA) segments and PA vascular smooth muscle cells (PAVSMCs) and PA endothelial cells (PAECs).
Methods: Segments and cells were incubated under hypoxic conditions for 2 hours (with or without enzyme inhibitors) and the formation of O2− measured spectrophotometrically. Protein expression was assessed using Western blotting and immunocytochemistry.
Results: Hypoxia promoted the formation of O2− in PA segments, PAVSMCs and PAECs, an effect inhibited by diphenylene iodonium and apocynin (NAD[P]H oxidase inhibitors). Hypoxia induced O2− formation was enhanced by inhibition of eNOS and augmented by endotoxin and cytokines and re-oxygenation. Hypoxia also promoted the expression of gp91phox and eNOS. In intact PA segments hypoxia induced the expression of nitrotyrosine and eNOS in the endothelium.
Conclusions: The simultaneous upregulation of NAD[P]H oxidase and eNOS in response to hypoxia in the PA results in the simultaneous formation of O2−, NO, and ONOO−. This may represent either a protective mechanism designed to counter the pro-oxidant effect of hypoxia or a novel pathological mechanism underlying the progression of acute respiratory distress syndrome (ARDS).
- AP-1, activator protein-1
- ARDS, acute respiratory distress syndrome
- DPI, diphenylene iodonium
- IL-1α, interleukin 1α
- LPS, lipopolysaccharide
- NADPH, reduced nicotinamide-adenine dinucleotide phosphate
- NF-κB, nuclear factor-kappa B
- NO, nitric oxide
- NOS, nitric oxide synthase
- O2−, superoxide
- ONOO−, peroxynitrite
- PAECs, pulmonary artery endothelial cells
- PAVSMCs, pulmonary artery vascular smooth muscle cells
- SOD, superoxide dismutase
- TNF-α, tumour necrosis factor-α
- TXA2, thromboxane A2
- nitric oxide
- hypoxia
- superoxide
- pulmonary artery
- acute respiratory distress syndrome (ARDS)
Statistics from Altmetric.com
- AP-1, activator protein-1
- ARDS, acute respiratory distress syndrome
- DPI, diphenylene iodonium
- IL-1α, interleukin 1α
- LPS, lipopolysaccharide
- NADPH, reduced nicotinamide-adenine dinucleotide phosphate
- NF-κB, nuclear factor-kappa B
- NO, nitric oxide
- NOS, nitric oxide synthase
- O2−, superoxide
- ONOO−, peroxynitrite
- PAECs, pulmonary artery endothelial cells
- PAVSMCs, pulmonary artery vascular smooth muscle cells
- SOD, superoxide dismutase
- TNF-α, tumour necrosis factor-α
- TXA2, thromboxane A2
Acute respiratory distress syndrome (ARDS), a severe form of acute lung injury, is a common complication in critically ill patients and is associated with significant morbidity and mortality.1–4 Although ARDS can be initiated by a number of causal factors including sepsis, shock, trauma and multiple transfusions, the pathological and clinical manifestations of the syndrome are very similar.1 Oxidative stress, in particular superoxide (O2−) formation, is involved in the aetiology of ARDS,1–4 a condition characterised by a rapid and time dependent worsening of intrapulmonary inflammation and hypertension.4 Apart from directly eliciting vasoconstriction,3 O2− reacts with nitric oxide (NO) to form peroxynitrite (ONOO−) and other reactive nitrogen species,5 effectively reducing NO bioavailabilty (fig 1). ONOO− is a double edged sword in that it is a vasodilator of pulmonary arteries (PAs),6 yet it is a potent pro-oxidant that can lead to tissue damage and worsening of inflammatory cascades.7 However, since NO is a vasodilator and inhibits adhesion molecule expression,8,9 the reduction of NO bioavailability may promote vasoconstriction and therefore pulmonary hypertension. A reduction in NO may also enhance the adhesion of leucocytes and platelets leading to augmented inflammation.8,9 In this context, endotoxin, tumour necrosis factor-α (TNF-α), interleukin-1α (IL-1α), and thromboxane A2 (TXA2), all promote O2− formation via the upregulation of NAD[P]H oxidase in isolated PA.10–12 Since these factors are all hallmarks of ARDS, it has been suggested that this may constitute a mechanism by which pulmonary hypertension and inflammation is promulgated and worsened in ARDS (fig 1).

Schematic representation of the pathological events leading to increased NADPH oxidase expression in ARDS. Inflammatory triggers including endotoxin firstly promote the adhesion of blood cells to the pulmonary arterial endothelium. These then release a battery of factors including cytokines and eicosanoids10,11 which upregulate the expression of NADPH oxidase, thereby increasing the endogenous formation of superoxide (O2−).10,11 O2− promotes inflammation and vasoconstriction in its own right but also reduces NO derived from eNOS to form reactive nitrogen species (RNS). Since NO is a potent endogenous inhibitor of blood cell adhesion and activity and is a vasodilator, this reduction augments the inflammatory and hypertensive sequelae of ARDS. Since NO also suppresses the expression of NAD[P]H oxidase,12 this reduction of NO results in further augmentation of NAD[P]H oxidase expression and activity. By contrast, cytokines also upregulate eNOS expression which may have two consequences: a negation of the upregulation of NAD[P]H oxidase or a greater formation of RNS.9 Finally, in the early stages of ARDS, these inflammatory events lead to tissue hypoxia. Since hypoxia upregulates many other proteins including NOS, it is reasonable to suggest that hypoxia may upregulate the expression of NAD[P]H oxidase which, in turn, would worsen and amplify ARDS.
By contrast, endogenous NO derived from endothelial NO synthase (eNOS) appears to play a role in removing excess O2− in pulmonary arterial tissue and cells which have been challenged with endotoxin, cytokines, and eicosanoids.10 NO is also a potent inhibitor of NAD[P]H oxidase expression and activity in pulmonary arterial endothelium.12 Jernigan et al13 also showed that the inhibition of NOS leads to greater generation of reactive oxygen species (ROS) in lungs in experimental hypoxia. The negation of NO availability by O2− may therefore render pulmonary vasculature even more susceptible to oxidative attack since the inhibitory effect of NO on NAD[P]H oxidase upregulation would be negated by O2− (fig 1).
Another important aetiological component of pulmonary arterial disease is that of oxygenation status—namely, tissue hypoxia and re-oxygenation.1–3 In ARDS, in particular, inflammation can lead to the rapid development of acute alveolar hypoxia4 which, apart from directly eliciting vasoconstriction,14 also increases O2− generation in vascular tissue.14,15 Hypoxia acutely promotes O2− generation from disparate intracellular sources that include xanthine dehydrogenase oxidase,16 mitochondrial electron transport chain,17 and NAD[P]H oxidase itself.18 Furthermore, the main supportive treatment of the failing respiratory system in ARDS is mechanical ventilation which delivers high doses of oxygen and a continuous level of pressure (positive end-expiratory pressure) to the damaged lungs.19 However, this treatment in effect creates a severe hypoxia/re-oxygenation scenario, itself associated with increased O2− formation.5,20
It is also now well established that hypoxia promotes the expression of a wide number of pro-inflammatory genes including NOS.21 It is not known, however, whether hypoxia influences the expression of NAD[P]H oxidase. Given the potential pathogenic role of NAD[P]H oxidase in promoting acute lung injury and ARDS through O2− generation, it is important to determine whether hypoxia influences the expression of NAD[P]H oxidase. Such an event would further amplify oxidative stress-mediated pathology in acute lung injury and ARDS. To investigate the relationship between hypoxia and NAD[P]H oxidase, the acute effect of hypoxia (alone and interactively with endotoxin and cytokines) on O2− formation and the expression of gp91phox (active catalytic subunit of NAD[P]H oxidase10) in intact pig PA segments and vascular smooth muscle cells (PAVSMCs) and endothelial cells (PAECs) derived from the artery were therefore investigated. The source of O2− formation was determined using a range of relevant enzyme inhibitors.10–12 The role of the endothelium and of eNOS in mediating the effects of hypoxia on O2− formation was also studied with NOS inhibitors, the expression of eNOS, and the formation of nitrotyrosine (an index of the reaction between O2− and RNS7). Finally, the effect of re-oxygenation on hypoxia mediated O2− formation (alone and interactively with endotoxin and cytokines) was investigated.
METHODS
Materials
Dulbecco’s minimum essential medium (DMEM) and other tissue culture reagents were purchased from GibcoBRL (Paisley, Scotland). Endothelial cell growth medium was purchased from PromoCell (Heidelberg, Germany). IL-1α and TNF-α were purchased from R&D Systems (Abingdon, UK). BCA-protein assay kit was purchased from Pierce (Rockford, Illinois, USA). A specific monoclonal antibody against eNOS was purchased from Transduction Laboratories (Oxford, UK) and nitrated tyrosine from Upstate Biotechnology (Buckinghamshire, UK). Secondary antibodies were from Dako Ltd (Ely, Cambridgeshire, UK). A specific monoclonal human antineutrophil gp91phox antibody (MoAb 48) was a kind gift from Professor D Roos (CLB, Amerstdam, The Netherlands).22 Nitric oxide metabolite kits were purchased from Calbiochem (UK). All other chemicals were from Sigma Chemical Co (Poole, Dorset, UK).
Dissection and preparation of pulmonary arteries
White Landrace male pigs (35 kg) were anaesthetised with ketamine hydrochloride (10 mg/kg; Ketaset Injection, Fort Dodge Animal Health, Southampton, UK) and inhaled oxygenated halothane, exsanguinated, and the lungs removed. All animal procedures were carried out according to Home Office regulations for which we hold an appropriate licence. Pulmonary arteries (3–4 mm diameter) were dissected out and placed in DMEM with Glutamax-1. The arteries were then cut into 2–3 mm2 segments for experimentation. When required, the endothelium was removed by gently rubbing the lumenal surface with a cotton wool bud.
Cell culture of PAVSMCs and PAECs
Pulmonary artery vascular smooth muscle cells (PAVSMCs) and endothelial cells (PAECs) were prepared according to previously published methods. PAECs were grown in endothelial cell growth medium at 37°C in a 95% air/5% CO2 incubator (Heraeus, Hera Cell, Kandro Laboratory Products, Germany). PAVSMCs were maintained in DMEM (supplemented with 10% fetal calf serum) at 37°C in a 95% air/5% CO2 incubator. Subconfluent cultures of pulmonary VSMCs were growth arrested by washing in sterile phosphate buffered saline (PBS) and incubating in quiescing medium (serum free DMEM supplemented with 0.5% lactalbumin hydrolysate) for 72 hours. Cultures were then incubated in fresh serum free medium supplemented with the factor(s) under investigation.
Measurement of superoxide
The measurement of O2− release by arterial segments or cultured cells was performed by detection of ferricytochrome c reduction. Following incubation, arterial segments or cells were rinsed three times with PBS and equilibrated in DMEM without phenol red for 10 minutes at 37°C in a 95% air/5% CO2 incubator. 20 μM horseradish cytochrome c with or without 500 U/ml copper-zinc superoxide dismutase (SOD) was added to the segments and incubated at 37°C in a 95% air/5% CO2 incubator for an hour. The final volume of the reaction mixture was 0.5 ml per well. After 1 hour the reaction medium was removed and maximum rate of reduction of cytochrome c was determined at 550 nm on a temperature controlled anthos Lucy 1 spectrometer (Lab-tech International, Ringmer, East Sussex, UK) and converted to nmoles O2− using ΔE550 nm = 21.1 mM/cm as the extinction coefficient for (reduced-oxidised) cytochrome c. The reduction of cytochrome c that was inhibitable with SOD reflected actual O2− release. Segments were blotted, dried and weighed, and data were expressed as nmol O2−/mg tissue/h. Cells were rinsed in PBS, lysed with 0.1% v/v Triton-X100, and total protein content measured using BCA-protein assay kit.
Effect of hypoxia on O2− generation in pulmonary arterial tissue and cells
PA segments (with and without endothelium), PAECs, or PAVSMCs were placed in multi-well plates as described above and ferricytochrome c (±SOD) added to the oxygen deprived medium. The plates were then transferred to a chamber which was flushed with 95% N2/5% CO2 for 10 minutes, then tightly sealed and the cells or segments incubated at 37°C for 2 hours. O2− was measured as described above. In order to determine the source of O2−, PA segments or cultured cells were pre-incubated for 2 hours with (1) diphenylene iodonium (DPI; 10 μM; a NADPH oxidase inhibitor), (2) apocynin (1 μM; a selective NADPH oxidase inhibitor), (3) allopurinol (100 μM; a xanthine oxidase inhibitor), or (4) rotenone (10 μM; mitochondrial electron transfer chain (METC) inhibitor). In order to study a possible role for NOS-derived NO in nullifying O2− formation (O2−+NO = ONOO−), PA segments or cells were pre-incubated for 2 hours with: Nw-nitro-l-arginine methyl ester hydrochloride (l-NAME; NOS inhibitor; 100 μM); N5-(1-iminoethyl)-ornithine (l-NIO; eNOS inhibitor; 10 μM) or l-N6-(1-iminoethyl)-lysine-HCl (l-NIL; iNOS inhibitor; 10 μM). PA segments and cells were then subjected to hypoxia for a further 2 hours and O2− measured by ferricytochrome c reduction assay as above.
Effect of hypoxia on gp91phox and eNOS protein expression
Following incubation for 2 hours under hypoxic conditions as described above, PAVSMCs or PAECs were rinsed in PBS and lysed with Tris buffer (50 mM, pH 7.4) containing 1% v/v Triton X-100, EDTA (10 mM), PMSF (1 mM), pepstatin (0.05 mM) and leupeptin (0.2 mM). Extracts were boiled at a 1:1 ratio with Tris (50 mM; pH 6.8 containing 4% w/v sodium dodecyl sulfate; 10% v/v glycerol; 4% v/v 2-mercaptoethanol; 2 mg/ml bromophenol blue). Samples of equal protein (100 μg) were loaded onto 12% Tris-glycine sodium dodecyl sulfate gels and separated by electrophoresis. After transfer to nitrocellulose, the blots were primed with either MoAb 48 (2.5 μg/ml final concentration) or anti-eNOS (1:2500 dilution). The blots were then incubated with goat anti-mouse antibody conjugated to horseradish peroxidase (1:2000 dilution) and developed by enhanced chemiluminescence (Amersham International). Rainbow markers (14–220 kDa; Amersham) were used for molecular weight determination.
Immunocytochemistry of eNOS and nitrated tyrosine
Immunocytochemical analysis of eNOS and nitrated tyrosine (NT) was carried out in selected samples of PA segments following 2 hour exposure to hypoxia. The segments were then snap frozen in liquid nitrogen and stored at −80°C. Cryostat sections (8 μm) were prepared and fixed in acetone for 10 minutes. The endogenous peroxide activity was inhibited with 1.2% H2O2 in methanol for 30 minutes. Sections were then blocked with goat serum, drained, and incubated with monoclonal antibodies against eNOS (1:200 dilution) and NT (0.3 μg/ml) overnight at 4°C. After washing in PBS, the sections were treated with either biotinylated goat anti-rabbit (NT; 1:200 dilution) or biotinylated goat anti-mouse (eNOS; 1:200 dilution) for 1 hour at room temperature, washed, and further treated for an hour with ExtrAvidin-Peroxidase (1:200 dilution). The bound antibody was visualised by addition of 0.05% diaminobenzidine (DAB, Dako Ltd) and 0.03% hydrogen peroxide in PBS which formed an insoluble brown precipitate (positive staining). The nuclei were counterstained using Mayer’s haematoxylin, dehydrated and mounted. Control slides (an irrelevant isotype matched antibody in place of the primary antibody) were prepared to test the specificity of staining.
Effect of LPS, IL-1α and TNF-α on hypoxia induced O2− formation
In order to determine the optimal time course and concentrations of LPS and cytokines for studying O2− release, PAVSMCs and PAECs were incubated with lipopolysaccharide (LPS; E coli; 026:B6; Sigma Chemical Co), human recombinant IL-1α (R&D Systems), or human recombinant TNF-α (R&D Systems) for different times and concentrations at 37°C in a 95% air/5% CO2 incubator. O2− release was measured by ferricytochrome c assay. To investigate the combined effects of hypoxia and cytokines on O2− generation, PA segments (±endothelium), PAVSMCs, or PAECs were pre-incubated with LPS (1 μg/ml; E coli; 026:B6), IL-1α (10 ng/ml), or TNF-α (10 ng/ml) for 16 hours. The cells and tissues were then incubated for 2 hours under hypoxic conditions and O2− release measured by ferricytochrome c reduction assay described above.
In order to determine the effect of re-oxygenation, PAVSMCs or PAECs were subjected to hypoxic conditions for 2 hours (±pre-incubation for 16 hours with LPS, IL-1α or TNF-α). The segments and cells were transferred from the hypoxic chamber to an incubator gassed with 5% CO2/95% O2 and equilibrated to normal atmospheric pressure for 1 hour. The formation of O2− was then measured by ferricytochrome c reduction assay.
Nitrite measurements
Nitric oxide generated by PAECs in response to hypoxia were measured as nitrite concentration using an assay kit based on the Griess reaction.23 Cells were cultured and exposed to hypoxic conditions described above in multi-well plates. At the end of the incubation the supernatants were removed and centrifuged to remove cell debris. Nitrate in the clarified culture supernatants was converted to nitrite by nitrate reductase. The nitrite was then reacted with sulphanilamide and N-(1-naphthyl) ethylenediamine and detected chromogenically by measuring the optical density at 540 nm with a spectrophotometer. A standard curve was calibrated using sodium nitrite at a concentration from 0–25 μM and the concentration of nitrite calculated by linear regression.
Data analysis
The data were investigated for normality by inspecting histograms and by applying the Kolmogorov-Smirnov test (automatically applied by Sigma Stat™ as part of the procedure for producing ANOVA results). In all cases the data did not deviate sufficiently from normality to warrant non-parametric statistics. Data were expressed as mean (SE). One-way and two-way analysis of variance (ANOVA) was used to determine statistical significance. Two-way ANOVA tests were employed where two conditions existed, such as with and without endothelium and the presence and absence of hypoxia (both with and without drugs), and one-way ANOVA was used when comparing effects of drug treatments with untreated controls.
RESULTS
Effect of hypoxia on O2− generation in pulmonary arterial tissue and cells
Hypoxia of 2 hours duration induced O2− formation from intact PA segments, PAECs, and PAVSMCs (fig 2). In intact PA segments, the removal of the endothelium reduced the formation of O2−, indicating that the endothelium contributes significantly to overall O2− output (fig 2A).

Effect of diphenylene iodonium (DPI; 10 μM; an NADPH oxidase inhibitor), apocynin (1 μM; a selective NADPH oxidase inhibitor), allopurinol (100 μM; a xanthine oxidase inhibitor), or rotenone (10 μM; mitochondrial electron transfer chain inhibitor) on hypoxia (2 hour duration) induced superoxide (O2−) formation in (A) intact porcine pulmonary artery segments with [+E] or without [−E] endothelium, (B) porcine pulmonary artery endothelial cells (PAECs), and (C) porcine pulmonary artery vascular smooth muscle cells (PAVSMCs) compared with controls (normoxic for 2 hours). Each point is the mean (SE) value, n = 6. *p<0.001, hypoxic tissues or cells v controls; †p<0.001, drug treated v untreated hypoxic tissues or cells; ‡p<0.001, with endothelium v without endothelium (in PA segments (A) only).
In PA segments (both with and without endothelium) and cultured PAVSMCs and PAECs, O2− formation induced by hypoxia of 2 hours’ duration was inhibited by DPI, apocynin, allopurinol and rotenone (fig 2), indicating that NADPH oxidase, xanthine oxidase, and components of mitochondrial electron transport chain mediate the effects of hypoxia on O2− formation.
l-NAME (100 μM) and the eNOS specific inhibitor l-NIO (10 μM) enhanced the production of O2− radicals in both PA segments (with intact endothelium) and PAECs subjected to hypoxia for 2 hours (fig 3). In contrast, the iNOS inhibitor l-NIL (10 μM) and l-NAME had no significant effect on hypoxia stimulated O2− release from endothelium denuded PA segments (fig 3), indicating that iNOS plays a minimal role in mediating these effects of hypoxia. These results indicate that eNOS derived NO may play a part in quenching hypoxia induced O2− radicals released from PA segments.

Effect of the NOS inhibitors l-nitroarginine methyl ester (l-NAME; 100 μM), N5-(1-iminoethyl)-ornithine (l-NIO; 10 μM), and l-N6-(1-iminoethyl)-lysine-HCl (l-NIL; 10 μM) on superoxide (O2−) formation by (A) 2 mm diameter porcine pulmonary artery segments with [+E] or without [−E] endothelium and (B) porcine pulmonary artery endothelial cells (PAECs) compared with controls (normoxic for 2 hours). Each point represents the mean (SE) value; n = 6. *p<0.001, drug treated hypoxic tissues or cells v untreated hypoxic tissue or cells.
Effect of hypoxia on gp91phox and eNOS protein expression and ONOO− formation
In cultured PAECs and PAVSMCs, hypoxia of 2 hours’ duration increased gp19phox protein expression as assessed by Western blotting (fig 4A). In cultured PAECs, hypoxia of 2 hours’ duration increased eNOS protein expression as assessed by Western blotting (fig 4B). Nitric oxide levels (measured as nitrite concentration) in PAEC culture supernatants were consistent with the upregulation of eNOS expression measured by immunoblotting. Thus, 2 hours of hypoxia significantly increased nitrite production by PAEC (9.90 (0.84) μM/mg protein) compared with normoxia (5.46 (0.53) μM/mg protein).

Effect of hypoxia on NAD[P]H oxidase and eNOS protein expressions in pig pulmonary arteries. (A) Expression of NAD[P]H oxidase in PAECs and PAVSMCs measured by Western analysis using a monoclonal antibody directed against the gp-91phox subunit of human neutrophil NADPH oxidase (MoAb 48). The bands detected are the 91 kDa for the heavily glycosylated form of gp-91phox and the 66 kDa for the less glycosylated form of gp-91phox. Pig neutrophil lysates served as positive control. (B) eNOS protein expression in PAECs measured by Western blotting using monoclonal antibodies raised against mouse eNOS. Human aortic endothelial lysate was used as positive control and the band detected is 140 kDa for eNOS. The bottom panels show the representative blots and the top panels the results of the densitometric analyses of six blots (expressed as relative optical density (OD)/mm2). *p<0.001, hypoxic v control values.
Immunocytochemistry confirmed that eNOS is upregulated in PAECs following a 2 hour incubation of intact PA segments under hypoxic conditions (fig 5D). Nitrated tyrosine was located principally in the endothelium following incubation of intact PA segments for 2 hours under hypoxic conditions, indicating that both NO and O2− are present at high concentrations in this region (fig 5H). Some immunoreactivity was also detected in the media of hypoxia treated PA segments (fig 5F).

Distribution of immunoreactive nitrated tyrosine (NT) and the expression of eNOS in pig pulmonary arteries. Freshly prepared pulmonary arterial segments were treated with vehicle (A, E), with 2 hour hypoxia (B, F), without the relevant primary antibody (C, G), or with control IgGs (D, H). Frozen sections of these segments were then immunostained for either eNOS (A–D) or nitrated tyrosine (E–H). Dark brown staining represents positive staining. The endothelium is indicated by arrows.
Effect of LPS, IL-1α and TNF-α on O2− formation
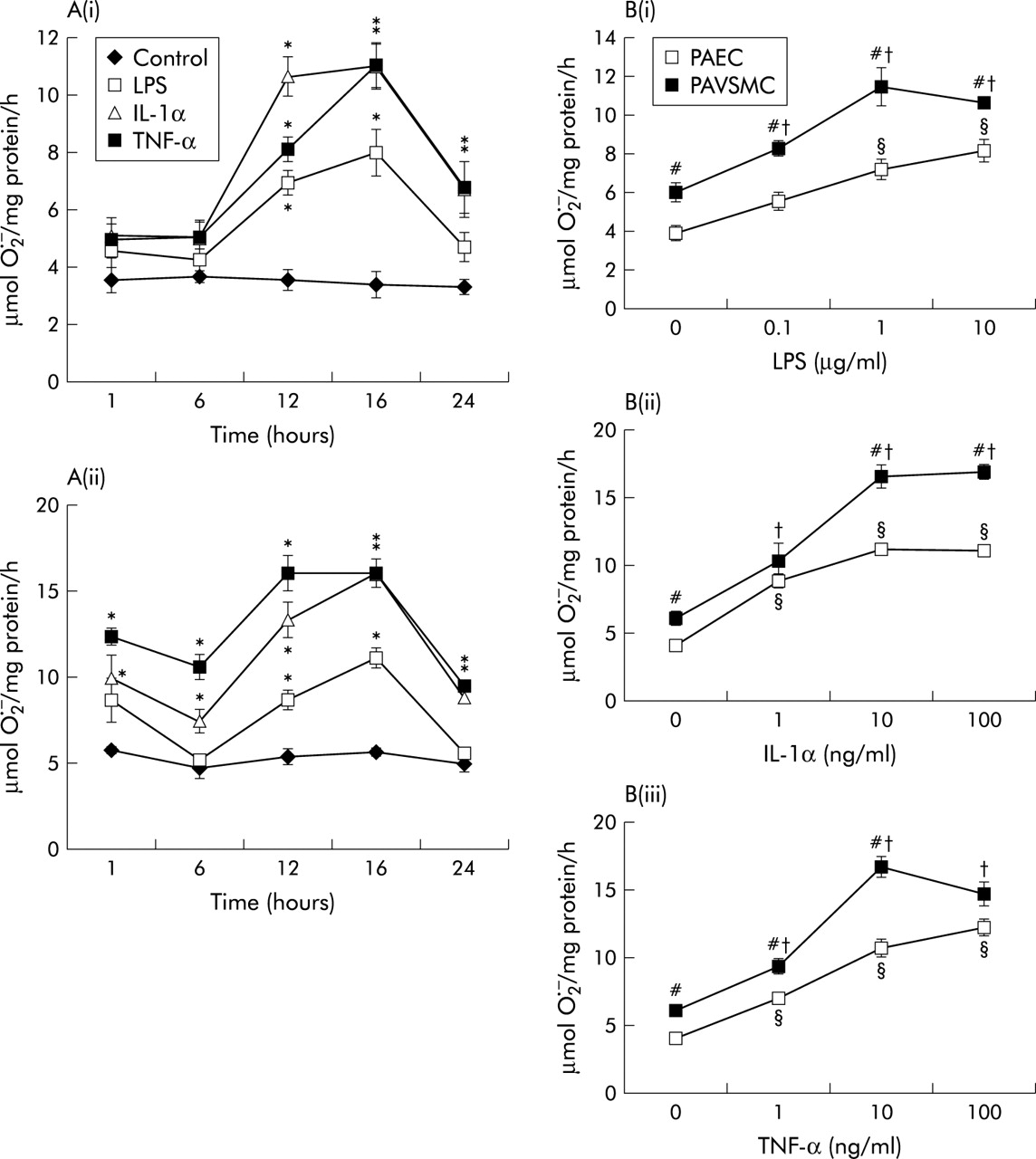
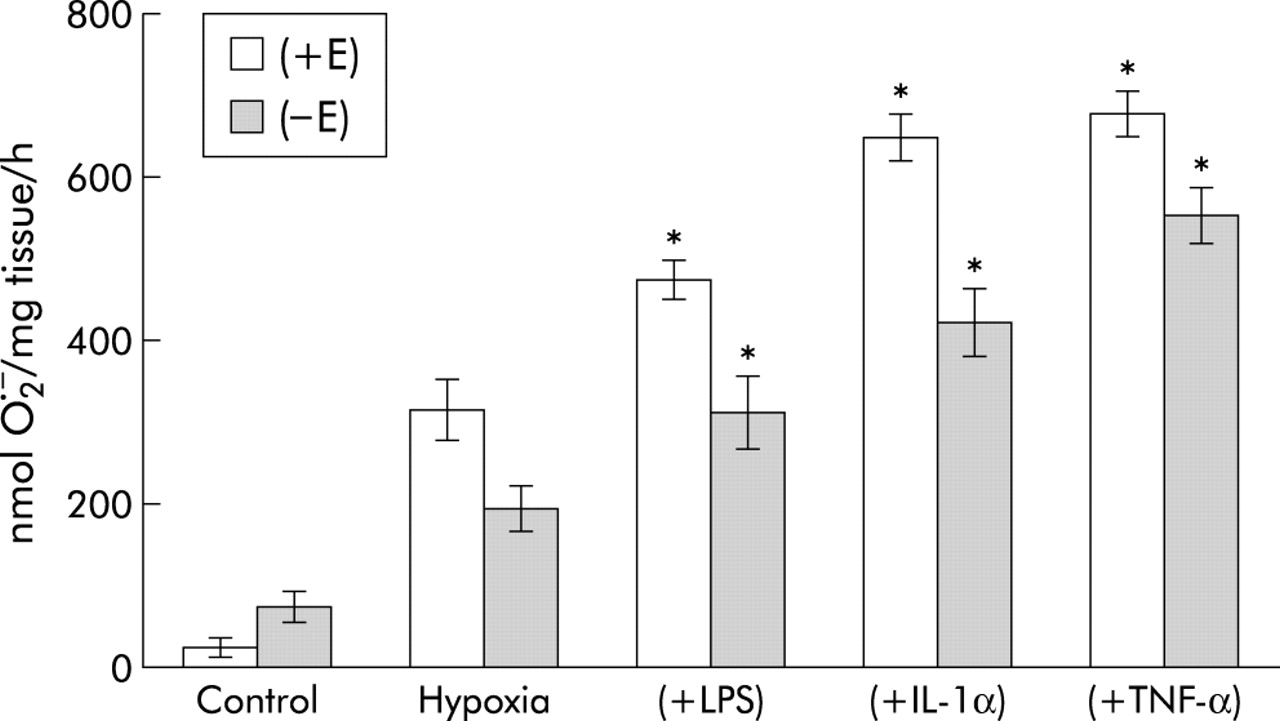
LPS (1 μg/ml) and TNF-α and IL-1α (both 10 ng/ml) augmented O2− release from PAECs and PAVSMCs in a time-dependent and dose-dependent manner (fig 6A and B). TNF-α and IL-1α promoted significantly greater amounts of O2− than LPS (fig 6A). Overall, PAVSMCs generated more O2− than PAECs per mg protein (fig 6B). For subsequent studies, a 16 hour time course and concentrations of 1 μg/ml LPS, 10 ng/ml IL-1α, and 10 ng/ml TNF-α were therefore used. Pre-incubation for 16 hours with LPS, IL-1α and TNF-α further augmented O2− formation by both intact PA segments as well as endothelium denuded segments (fig 7).

(A) Time course of effects of lipopolysaccharide (LPS; 1 μg/ml), interleukin 1α (IL-1α; 10 ng/ml) and tumour necrosis factor-α (TNF-α; 10 ng/ml) on superoxide dismutase (SOD) inhibitable O2− formation by (i) pulmonary arterial endothelial cells (PAECs) and (ii) pulmonary arterial vascular smooth muscle cells (PAVSMCs). (B) Concentration-response curves for (i) LPS, (ii) IL-1α, and (iii) TNF-α on SOD inhibitable O2− formation by PAECs and PAVSMCs following a 16 hour incubation. Each point represents a mean (SE) value; n = 6. *p<0.001, treated v controls at each time point; §p<0.001, responses of LPS or cytokines v zero (PAEC); †p<0.001, responses of LPS or cytokines v zero (PAVSMC); #p<0.001, PAVSMCs v PAECs at each concentration of cytokine or LPS.

Effect of 2 hour hypoxia following 16 hour pre-incubation with lipopolysaccharide (LPS; 1 μg/ml), interleukin 1α (IL-1α; 10 ng/ml), or tumour necrosis factor-α (TNF-α; 10 ng/ml) on O2− formation in pulmonary arterial segments with [+E] or without [−E] endothelium. Data are presented as mean (SE) values; n = 6. p<0.001, various combinations v hypoxia alone.
Effect of re-oxygenation on hypoxia induced O2− formation
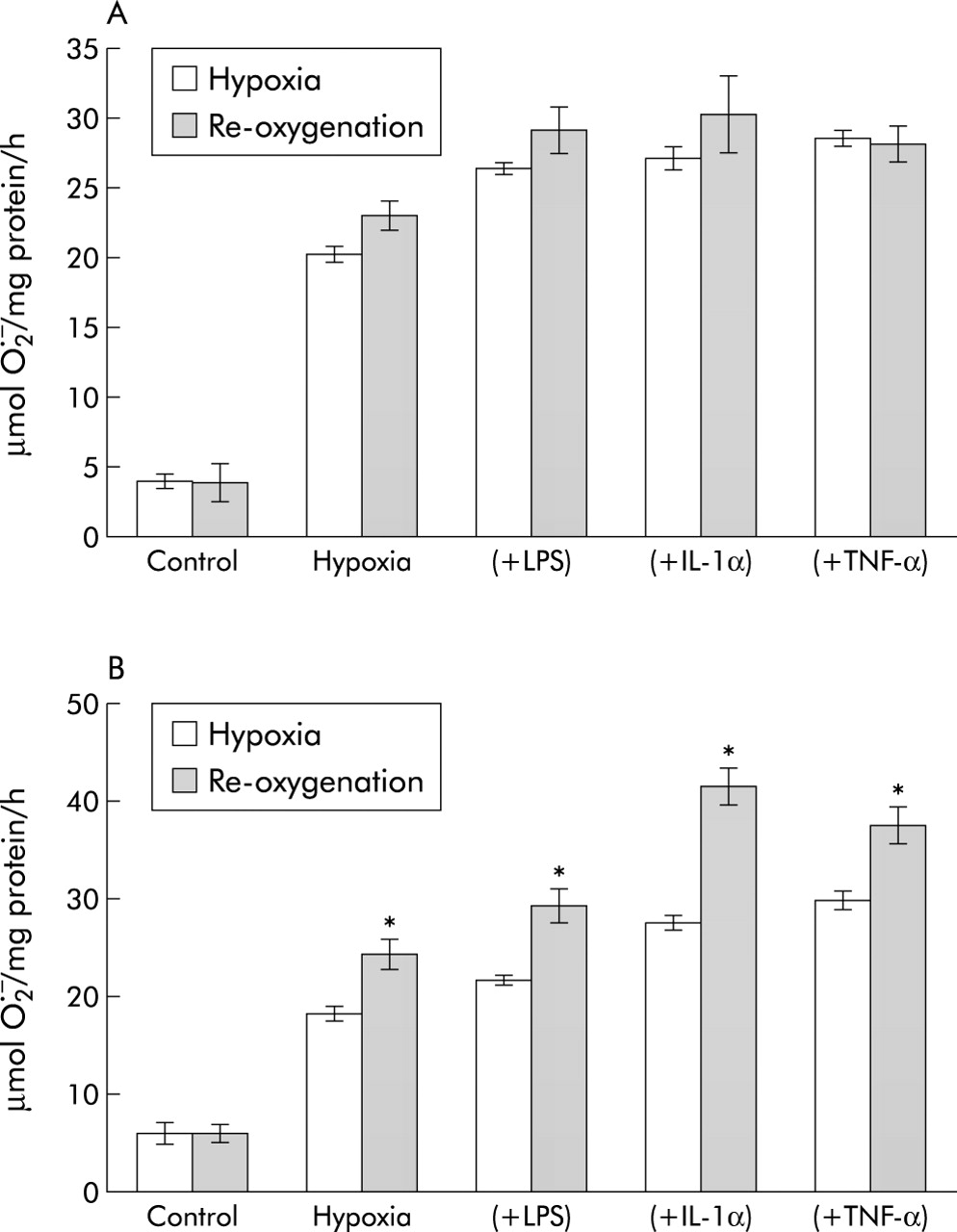
Re-oxygenation for 1 hour following 2 hour hypoxia (alone or after 16 hour pre-incubation with LPS, IL-1α or TNF-α) increased O2− generation from PAVSMCs but had no significant effect on hypoxic PAECs (fig 8).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of re-oxygenation with 95% O2 for 1 hour on O2− formation by (A) PAECS and (B) PAVSMCs following 2 hour hypoxia (with or without 1 μg/ml LPS; 10 ng/ml IL-1α, or 10 ng/ml TNF-α). Data represent mean (SE) values; n = 6. *p<0.001, re-oxygenated v non re-oxygenated cells.
DISCUSSION
Acute tissue hypoxia is an established component of the pathological sequelae of ARDS.1–5 Hypoxia is thought to be elicited in part by cytokines and other release factors from inflammatory and blood cells (as well as endotoxin in ARDS precipitated by sepsis) that accumulate in the lungs in acute lung injury.1,4,10–12 This study was undertaken to determine the effect of acute and severe hypoxia on pulmonary arterial expression of NAD[P]H oxidase and eNOS, since O2− and NO status have been implicated in the aetiology of pulmonary hypertension and ARDS.
As in previous reports, the present study shows that acute hypoxia increases O2− formation in intact PAs and cultured PAVSMCs and PAECs.24–28 O2− formation was inhibited by DPI (an NAD[P]H oxidase inhibitor), rotenone (a mitochondrial electron transport chain inhibitor), and allopurinol (a xanthine oxidase inhibitor). The inhibition of NAD[P]H oxidase by DPI has been shown to inhibit both hypoxia induced O2− production and vasoconstriction in isolated PAs.25,26 Xanthine oxidase activation has also been shown to play a role in hypoxia mediated O2− formation.27 During acute hypoxia the respiratory electron transport chain becomes reduced and the reduced state favours O2− production.28 Thus, in keeping with other studies, acute hypoxia promotes the generation of O2− in the PA from three sources: NAD[P]H oxidase, xanthine oxidase, and mitochondria.
In this study acute hypoxia also rapidly upregulated the expression of gp91phox, an active catalytic subunit of NAD[P]H oxidase,18 in both PAECs and PAVSMCs which, to our knowledge, is a novel observation. However, previous studies are consistent with this finding since hypoxia promotes those intracellular events that are also associated with the upregulation of NAD[P]H oxidase. For example, protein kinase C (PKC), nuclear factor-κB (NF-κB), and redox-sensitive transcription factor activator protein-1 (AP-1) are activated in bovine pulmonary arterial smooth muscle cells during hypoxia,29,30 and PKC, NF-κB and AP-1 mediate the expression of NAD[P]H oxidase.18
l-NAME (a non-specific NOS inhibitor) and inhibition of eNOS augmented O2− formation in PAECs and intact PAs, indicating that endogenous NO derived from eNOS has a role in removing excess O2−. In turn, acute hypoxia rapidly induced eNOS protein expression in PAECs and eNOS immunoreactivity in intact PAs, which is consistent with previous reports that hypoxia promotes an increase in eNOS protein.31 Nitrotyrosine (an index of ONOO−) was also found to be co-localised with enhanced eNOS in the endothelium of intact PA segments in response to acute hypoxia. Nitrite levels (index of NO formation) were also reduced in cells subjected to hypoxia. Together, these data confirm that there is increased NO and O2− formation derived from eNOS and NAD[P]H oxidase, respectively, and that they react to form reactive nitrogen species in the endothelium, effectively lowering NO availability.32 Jernigan et al13 showed that the inhibition of NOS increased ROS levels in arteries from rats subjected to chronic hypoxia and in control rats, which is also consistent with our data. The mechanism by which hypoxia increases eNOS expression is not fully understood since a hypoxia responsive element has not yet been identified in the eNOS promoter. It has been reported, however, that hypoxia activates redox sensitive transcription factor AP-1 in PAECs, which may mediate the increase in eNOS expression.33
Since NO and O2− exert opposite effects on pulmonary arterial function,2,5 the simultaneous upregulation of eNOS with NAD[P]H oxidase in response to acute hypoxia would seem paradoxical. However, the upregulation of eNOS may constitute a protective mechanism designed to negate the pathological impact of O2−. Apart from removing O2− by direct chemical reactions, NO derived from eNOS also inhibits the expression of NAD[P]H oxidase.10,12 A central role for eNOS in protecting against hypoxia induced pathogenesis is supported by the observation that a loss of eNOS leads to increased acute hypoxic vasoconstriction and development of pulmonary hypertension.33,34 Although the formation of ONOO− may reduce O2− levels in the pulmonary vasculature and therefore protect against the progression of ARDS and pulmonary hypertension per se, ONOO− exerts many pathological effects that would contribute to the progression of ARDS. These effects include the promotion of endothelial cell apoptosis,35 increased adhesion molecule expression,36 the inhibition of prostacyclin synthase,37 and inactivation of SOD.38
Clinically, the benefits of inhaled NO in treating ARDS have proved ambivalent.4 Adverse effects include a life threatening “rebound” increase in pulmonary vascular resistance. NO has also been shown to cause methaemoglobinaemia and cellular apoptosis.39–41 These latter observations indicate therefore that the upregulation of eNOS may in fact be deleterious when accompanied by an increase in O2− formation. By contrast, in a recent multicentre clinical trial in which the NOS inhibitor 546C88 was studied for safety and efficacy in 797 patients with septic shock, it was found that the inhibitor caused a marked increase in mortality.42 This latter trial would seem to indicate that the protective effect of endogenous NOS, particularly eNOS, may outweigh the other negative effects of NO and the metabolites derived from its reaction with O2−. However, inhalational NO may still be deleterious due to the large intrapulmonary levels of NO elicited by this treatment.
In the present study the effect of hypoxia on O2− formation was enhanced by endotoxin, TNF-α, IL-1α, and TXA2, all key promoters of inflammation in ARDS.2,3,10–12 We have previously shown that these factors all increase O2− formation in the pig PA, PAVSMCs, and PAECs through the upregulation of NAD[P]H oxidase and not by effects on xanthine oxidase or mitochondrial respiration.10–12 These data indicate that hypoxia may augment pulmonary pathogenesis in ARDS through interactions with disparate inflammogenic factors released by activated leucocytes and by bacteria. It is also of interest that, as with hypoxia, endotoxin, TNF-α and IL-1α all promote the expression of eNOS in these tissues and cells.10,12 It is therefore tempting to speculate that the intracellular signalling pathways that mediate the expression of NAD[P]H oxidase and eNOS in response to hypoxia and inflammogens are shared or interlinked. Furthermore, re-oxygenation following hypoxia enhanced the formation of O2− in PAVMSCs but not PAECs in both the absence and presence of cytokines and endotoxin. Since eNOS is upregulated in PAECs but not PAVSMCs, and we have shown that eNOS derived NO removes O2−, this would readily explain why this re-oxygenation effect is more apparent in PAVSMCs than PAECS. This may be relevant since the main supportive treatment of the failing respiratory system in ARDS is mechanical ventilation which delivers high doses of oxygen and a continuous level of pressure.19 This treatment may create a hypoxia/re-oxygenation scenario which, as our data indicate, may result in increased O2− formation.4,19,20
To summarise, this study shows that hypoxia alone and in combination with inflammogens associated with ARDS augments the formation of O2−, an effect mediated by the upregulation of NAD[P]H oxidase expression. This effect is augmented even further by re-oxygenation following hypoxia in PAVSMCs. Since O2− elicits a plethora of events that promote inflammation—including the negation of NO bioavailability, vasoconstriction and adhesion molecule expression—these interactions may play an important part in the progression of ARDS. Although the simultaneous upregulation of eNOS with gp91phox may negate the impact of O2−, the studies on intact PA segments with endothelium in which eNOS was found to be increased, the overall output of O2− was still enhanced. Furthermore, the concomitant upregulation of eNOS and NAD[P]H oxidase also resulted in increased ONOO− formation at the endothelial level which may have implications in the progression of ARDS, since ONOO− exerts a numbers of powerful effects that would promote inflammation. Therapeutically, therefore, a reduction in oxidative stress, especially the quenching of O2−, appears to be a potentially effective strategy in treating ARDS. Indeed, several studies in patients with ARDS and clinical studies and experiments in animal models have indicated that antioxidants may be beneficial in reducing the impact of oxidant stress in ARDS and pulmonary hypertension.43–48
Acknowledgments
The authors thank Rosemary Greenwood, Research and Development Support Unit, United Bristol Health Care Trust, for statistical advice.
REFERENCES
Footnotes
-
↵* Joint first author.
-
This research was funded by the British Heart Foundation (grant number FS/2001041).