Article Text

Abstract
Background Defective rhinovirus (RV)-induced interferon (IFN)-β and IFN-λ production and increased RV replication have been reported in primary human bronchial epithelial cells (HBECs) from subjects with asthma. How universal this defect is in asthma is unknown. Additionally, the IFN subtypes induced by RV infection in primary HBECs have not been comprehensively investigated.
Objective To study RV induction of IFN-α, IFN-β and IFN-λ and RV replication in HBECs from subjects with atopic asthma and healthy controls.
Methods HBECs were obtained from subjects with asthma and healthy controls and infected with RV16 and RV1B, and cells and supernatants harvested at 8, 24 and 48h. IFN proteins were analysed by ELISA and IFN mRNA and viral RNA expression by quantitative PCR. Virus release was assessed in cell supernatants.
Results IFN-β and IFN-λ were the only IFNs induced by RV in HBECs and IFN-λ protein induction was substantially greater than IFN-β. Induction of IFN-λ1 mRNA by RV16 at 48h was significantly greater in HBECs from subjects with asthma; otherwise there were no significant differences between subjects with asthma and controls in RV replication, or in induction of type I or III IFN protein or mRNA.
Conclusions IFN-λ and, to a lesser degree, IFN-β are the major IFN subtypes induced by RV infection of HBECs. Neither defective IFN induction by RV nor increased RV replication was observed in the HBECs from subjects with well controlled asthma reported in this study.
- Airway Epithelium
- Asthma
- Viral infection
Statistics from Altmetric.com
Key messages
What is the key question?
-
What are the important subtypes of interferon (IFN) induced by rhinovirus infection in bronchial epithelial cells and can the findings of deficient rhinovirus-induced IFN production be replicated in new groups of subjects with asthma?
What is the bottom line?
-
IFN-β and IFN-λ were the only IFNs induced by rhinovirus in bronchial epithelial cells and induction was not different in subjects with asthma and healthy controls.
Why read on?
-
Defective rhinovirus-induced type I and III IFN production is not a universal feature of asthma and may be related to asthma control.
Introduction
Asthma exacerbations are the major cause of morbidity, mortality and healthcare expenditure in asthma.1 Rhinovirus (RV) infection is responsible for at least 50% of asthma exacerbations and, compared with people without asthma, those with asthma experience more severe and longer lasting lower respiratory tract symptoms following infection.2 ,3 In response to RV infection, cells produce antiviral interferons (IFNs) (type I: IFN-α and IFN-β and type III: IFN-λ1 and IFN-λ2/3)4 and defective RV-induced IFN production has been implicated in the increased susceptibility to RV infection in asthma. Deficient RV induction of IFN-β has been reported in primary human bronchial epithelial cells (HBECs) and IFN-λ in HBECs and bronchoalveolar lavage (BAL) cells.5–7 Since the initial reports, defective IFN induction in asthma has also been extended to include deficient RV-induced IFN-α and IFN-β from BAL cells8 and IFN-α from peripheral blood mononuclear cells (PBMCs) from children.9 Deficient IFN-α subtype 2 (IFN-α2) response to respiratory syncytial virus and Newcastle disease virus has been reported in PBMCs from adults and children with atopic asthma10 ,11 and deficient influenza A virus induced IFN-α in plasmacytoid dendritic cells from subjects with asthma.12 Deficient IFN has also been identified in PBMCs from pregnant women with asthma.13 Defective RV-induced IFN production was associated with virus load in vitro5 ,7 and virus load and markers of exacerbation severity in vivo,5 suggesting that defective IFN induction was likely to be important in RV-induced asthma exacerbations. Deficient RV-induced type I IFN induction in BAL cells has been associated with greater airway hyperresponsiveness and greater positivity on allergy skin prick testing8 and influenza A induced IFN-α secretion inversely correlated with serum IgE,12 suggesting defective IFN induction in asthma may be related to more severe disease.
Despite this, some investigators have failed to identify defective IFN induction in asthma. A small study of HBECs cultured from four subjects with asthma found significantly greater mean levels of IFN-β in those with asthma.14 Another study employing genome-wide transcriptional analysis of HBECs found no difference in RV or IFN expression in subjects with asthma when cultured HBECs were infected with RV1A.15 Differences in experimental technique and clinical characteristics of the subjects studied exist between these studies and the original reports, which could be responsible for the conflicting results.
Bronchial epithelial cells are the major cell type infected by RVs in the lower respiratory tract in vivo.16 ,17 Deficient IFN induction by RVs has also been reported in lower airway cells from subjects with chronic obstructive pulmonary disease (COPD)18 and cystic fibrosis.19 Replacing deficient IFN is a potential novel therapeutic approach in clinical development for acute exacerbations of all three diseases. Understanding which IFN subtypes are induced by RV infection of primary HBECs would have major implications in determining which IFN subtype might have the best therapeutic potential. However, the IFN subtypes induced by RV infection of HBECs are poorly understood. One single study investigated RV induction of all three IFN subtypes in HBECs4; however, this study did not detect any IFN protein production. Further, the cells studied came from only three poorly characterised subjects obtained commercially. Finally the cells had been stored frozen for long periods of time prior to being studied. Thus there is a need to investigate IFN subtype induction in cells from larger numbers of well characterised subjects and in cells that have not been frozen for long periods prior to being cultured.
This study aimed to investigate the IFN subtypes induced by RV infection of primary HBECs cultured directly from larger numbers of subjects with and without asthma and to determine whether the original findings of defective RV-induced IFN-β and IFN-λ production could be replicated in HBECs cultured from new groups of subjects with asthma and healthy controls.
Methods
Subject recruitment
Twenty-two subjects with asthma and 20 without asthma were recruited by newspaper advertisement. None were smokers or had had exacerbations or respiratory tract infections in the preceding 6 weeks. Asthma was defined in individuals with atopic disease as a physician's diagnosis of asthma with bronchial hyperresponsiveness or evidence of reversibility to salbutamol.8 Atopy was defined as one or more positive skin prick tests to common aeroallergens (see online supplementary methods). Subjects without asthma were non-atopic and had no previous relevant medical history and no bronchial hyperresponsiveness (table 1 and see online supplementary methods). All participants underwent clinical characterisation described in online supplementary methods. All subjects gave written informed consent.
Clinical characteristics of participants from whom human bronchial epithelial cell (HBEC) samples were successfully obtained
HBEC isolation and processing
HBECs were obtained by fibre optic bronchoscopy using a Keymed BF260 bronchoscope (Olympus, Essex, UK) using 5 mm sheathed endobronchial brushes (Olympus BC-202D-5010) in accordance with British Thoracic Society guidelines.20 Freshly brushed HBECs were seeded into hormonally supplemented bronchial epithelial growth medium (BEBM, Lonza) plus 5000 units penicillin and 5000 μg streptomycin. Cell culture was performed as described previously.7 Cells were confirmed as epithelial by cytokeratin 19 staining by immunofluorescence.21 Cells were seeded at passage 2 onto 24 well plates and cultured until 80% confluent before infection with RV.7
RV infection
RV16 and RV1B stocks were generated and titrated as previously described5 and HBECs were cultured ex vivo as a monolayer and infected with RV at a multiplicity of infection of 1 for 1h. Virus was then removed, cells washed three times and replaced by fresh medium and cell lysates and supernatants were harvested at 8, 24 and 48h. As negative controls, cells were treated with medium alone.
RNA isolation and quantitative PCR
Total RNA was extracted from cells using RNeasy MiniKit (Qiagen, Hilden, Germany) and used for cDNA synthesis by Omniscript RT kit (Qiagen). Reactions were analysed using an ABI 7000 TaqMan (ABI Foster City, California, USA) as previously described.8 Results were normalised to 18 s.8 For primer and probe sequences, see online supplementary table 1.
Virus titration
Virus titration was performed by tissue culture infectious dose 50 (TCID50). Serial 1/10 dilutions of cell supernatants were added to HeLa cells in replicates, incubated for 4 days and cyto-pathological effect recorded by blinded investigators.
IFN protein measurement
IFN-β (Invitrogen, California, USA), IFN-α (Invitrogen) and IFN-λ (R&D systems) release were measured by ELISA according to the manufacturer's instructions. IFN-λ ELISA detects mainly IFN-λ1, however there is approximately 25% cross reactivity with IFN-λ2/3. The sensitivities of each assay were 10 pg/mL.
Statistical analysis
Statistical analysis was performed using Graphpad prism (V.5.04; GraphPad Software, San Diego, California, USA). IFN and proinflammatory cytokine induction by RV1B and RV16 were compared between subjects with atopic asthma and those without atopic asthma using Mann–Whitney testing on each pair of groups corrected for multiple testing with Dunn's correction. A p value of <0.05 was considered significant. Data are presented as medians unless indicated otherwise.
p Values for clinical characteristics were performed using t tests or Mann–Whitney tests depending on normality of data. Fisher's exact test was used for sex breakdown.
Results
HBEC samples were obtained from 22 subjects with asthma and 20 without asthma. Due to problems experienced with HBEC culture (reasons for failure to grow are provided in the online supplement), successful culture samples were only obtained for 10 subjects with asthma and 12 without asthma. The clinical characteristics of these participants are shown in table 1.
IFN-λ and IFN-β are the only IFN proteins induced in HBECs by RV infection and RV-induced IFN-β and IFN-λ protein were not significantly different in HBECs from subjects with and without asthma
To investigate IFN subtypes induced in HBECs and to establish whether the previously reported defective RV-induced IFN-β and IFN-λ in HBECs from subjects with asthma was also present in HBECs cultured from this new group of subjects with asthma, type I and III IFN proteins were measured at 8, 24 and 48h in HBECs infected with RV16 and RV1B or incubated with medium alone.
IFN-α protein was not detectable in samples from any subject at any time point (data not shown) and IFN-β protein was only significantly induced by RV1B at 48h (figure 1A). RV16 did not significantly induce IFN-β or IFN-λ protein at any time point and RV1B did not significantly induce IFN-β or IFN-λ protein at 8 and 24h. Significant IFN-λ protein induction was only observed at 48h post RV1B infection (figure 1B). Levels of IFN-λ induced by RV1B at 48h (median 358 pg/mL) were substantially and significantly greater than those of IFN-β (median 22 pg/mL (p<0.001)). No difference was observed between subjects with and those without asthma in IFN-β or IFN-λ protein induction by RV1B at 48h (figure 1A,B).
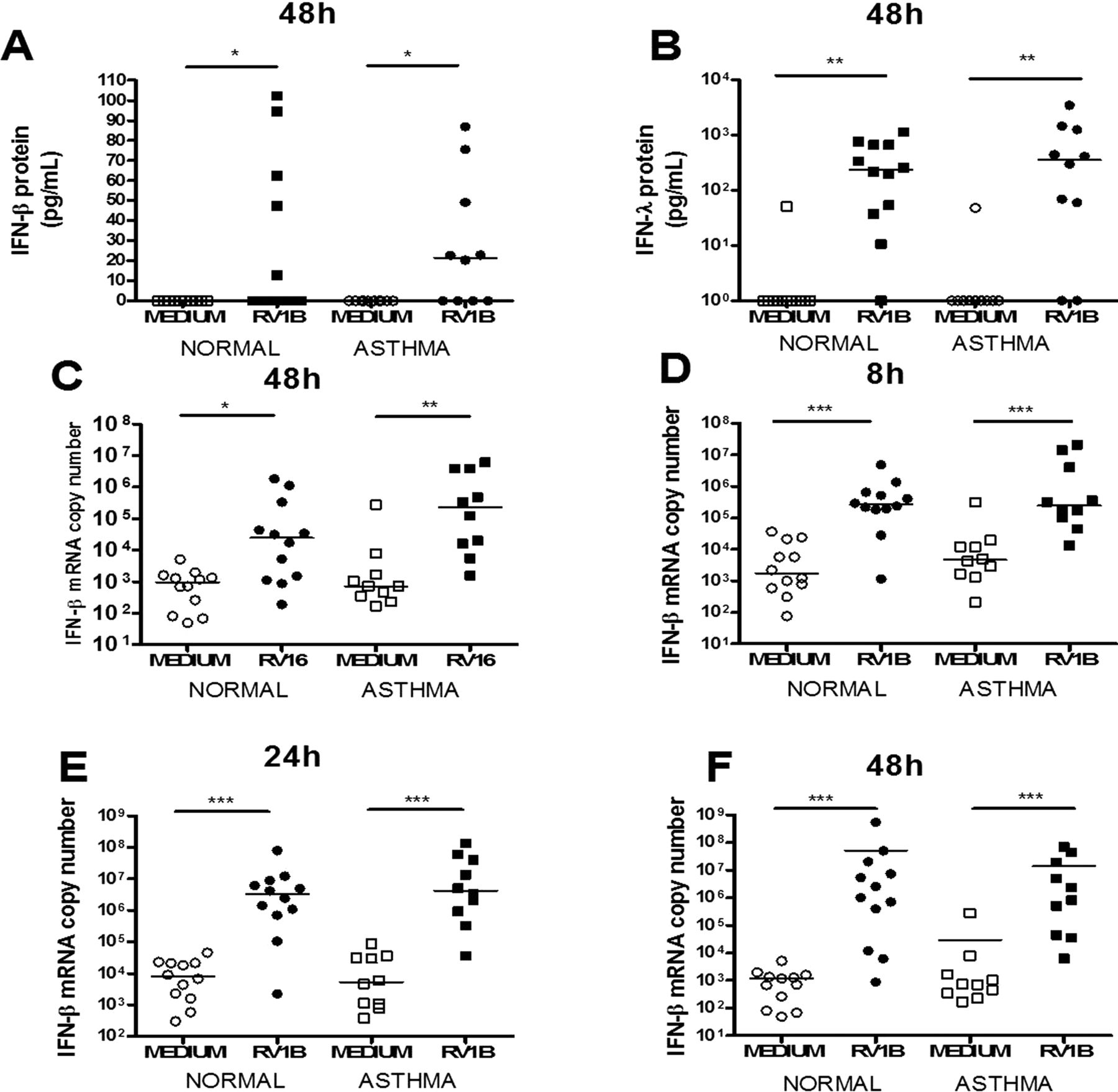
Rhinovirus (RV)-induced interferon (IFN)-β and IFN-λ protein and IFN-β mRNA induction in human bronchial epithelial cells (HBECs) was not different in subjects with asthma compared with healthy controls. IFN-β protein at 48h post RV1B (A), IFN-λ protein at 48h post RV1B (B), IFN-β mRNA at 48h post RV16 (C) and 8h (D), 24h (E) and 48h (F) post RV1B infection. Squares denote subjects with asthma, circles denote health controls. *p<0.05, **p<0.01 and ***p<0.001.
IFN-λ and IFN-β are the only IFN mRNAs induced in HBECs by RV infection and RV-induced IFN-β and IFN-λ mRNA was not significantly different in HBECs from subjects with and without asthma
To further study IFN subtypes induced by RV in HBECs, and to establish whether defective RV-induced type I and III IFN mRNA expression was present in HBECs in this group of subjects with asthma, IFN-α, IFN-β and IFN-λ mRNA expression was measured at 8, 24 and 48h in response to RV16 and RV1B.
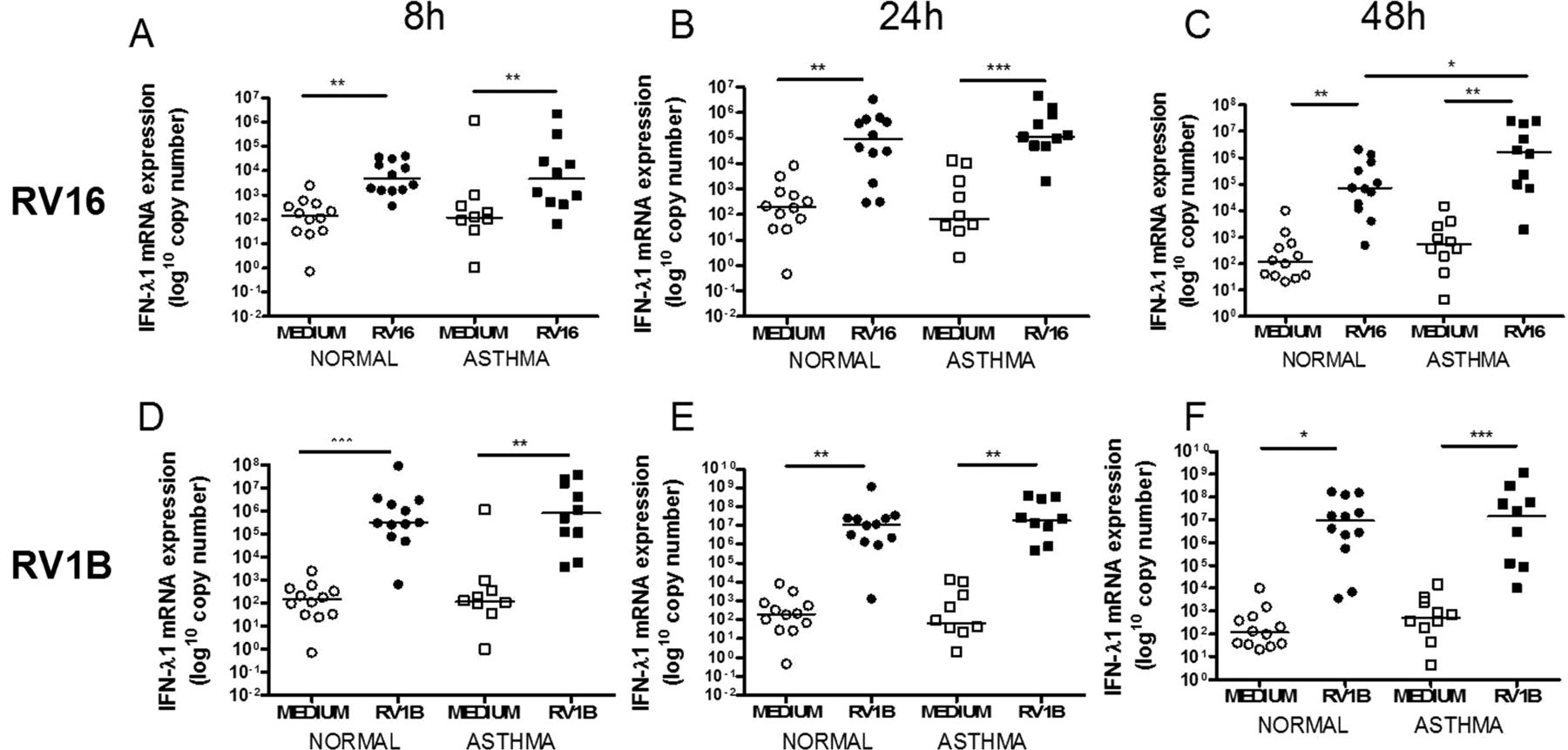
IFN-α mRNA was not induced by either virus (data not shown). IFN-β mRNA was significantly induced by RV16 only at 48h (figure 1C) and by RV1B at all time points (figure 1D–F). IFN-λ1 mRNA was induced by both viruses at all time points (figure 2) and IFN-λ2/3 mRNA was induced by RV16 at 8 and 24h in subjects with and without asthma and at 48h in those with asthma only (figure 3A–C). RV1B infection induced IFN-λ2/3 induction at 8, 24 and 48h (figure 3D–F). There was no significant difference between subjects with and without asthma for IFN-β, IFN-λ1 or IFN-λ2/3 mRNA induction after RV1B infection, however after RV16 infection there was significantly more IFN-λ1 mRNA in subjects with asthma at 48h (figure 2C). There were no significant differences in induction of IFN-β, IFN-λ1 or IFN-λ2/3 by either virus at any time point.
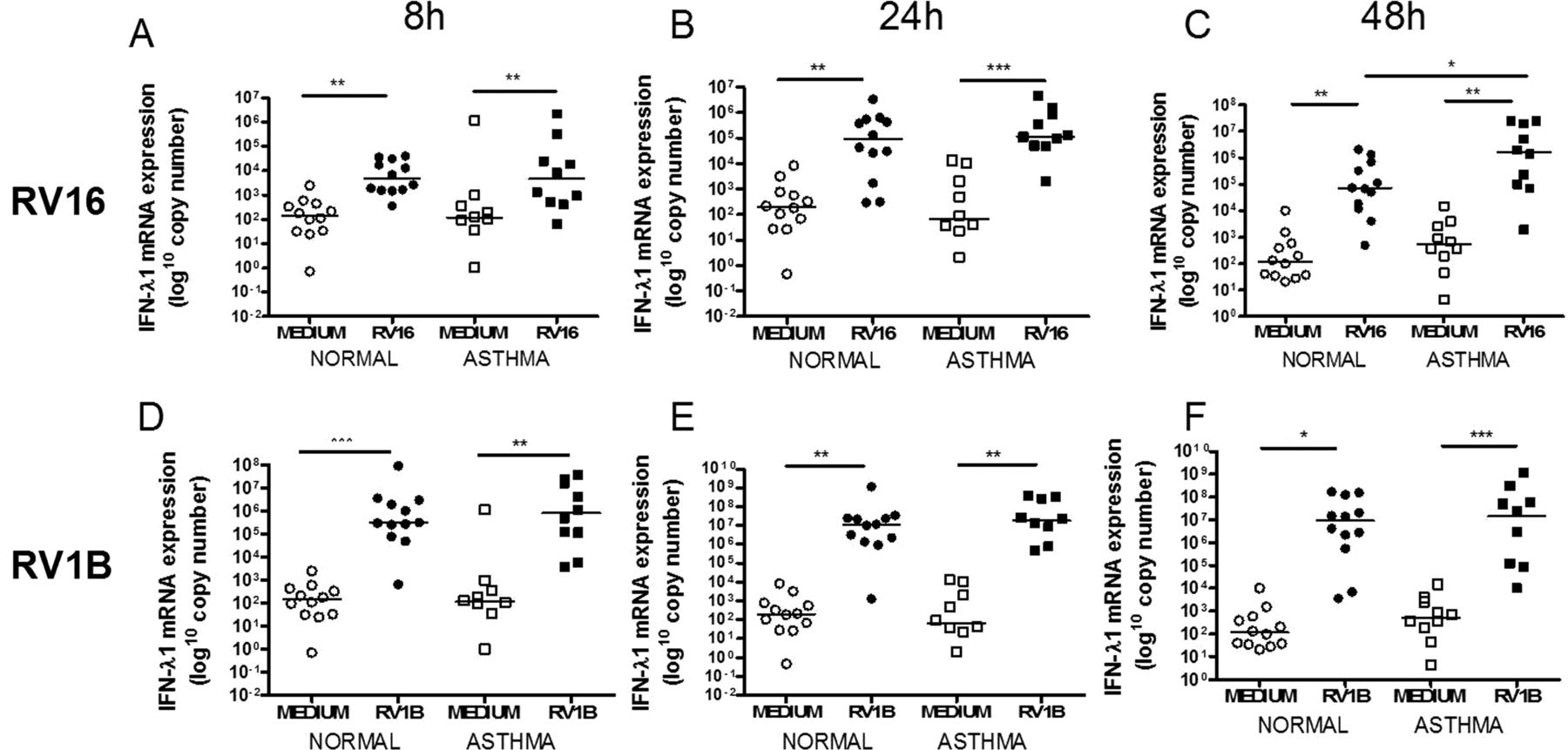
Interferon (IFN)-λ1 mRNA induction after rhinovirus (RV)-1B and RV16 was not different in subjects with asthma compared with healthy controls. IFN-λ1 mRNA at 8h (A), 24h (B) and 48h (C) post RV16 infection and 8h (D), 24h (E) and 48h (F) post RV1B infection. Squares denote subjects with asthma, circles denote healthy controls. *p<0.05, **p<0.01 and ***p<0.001.
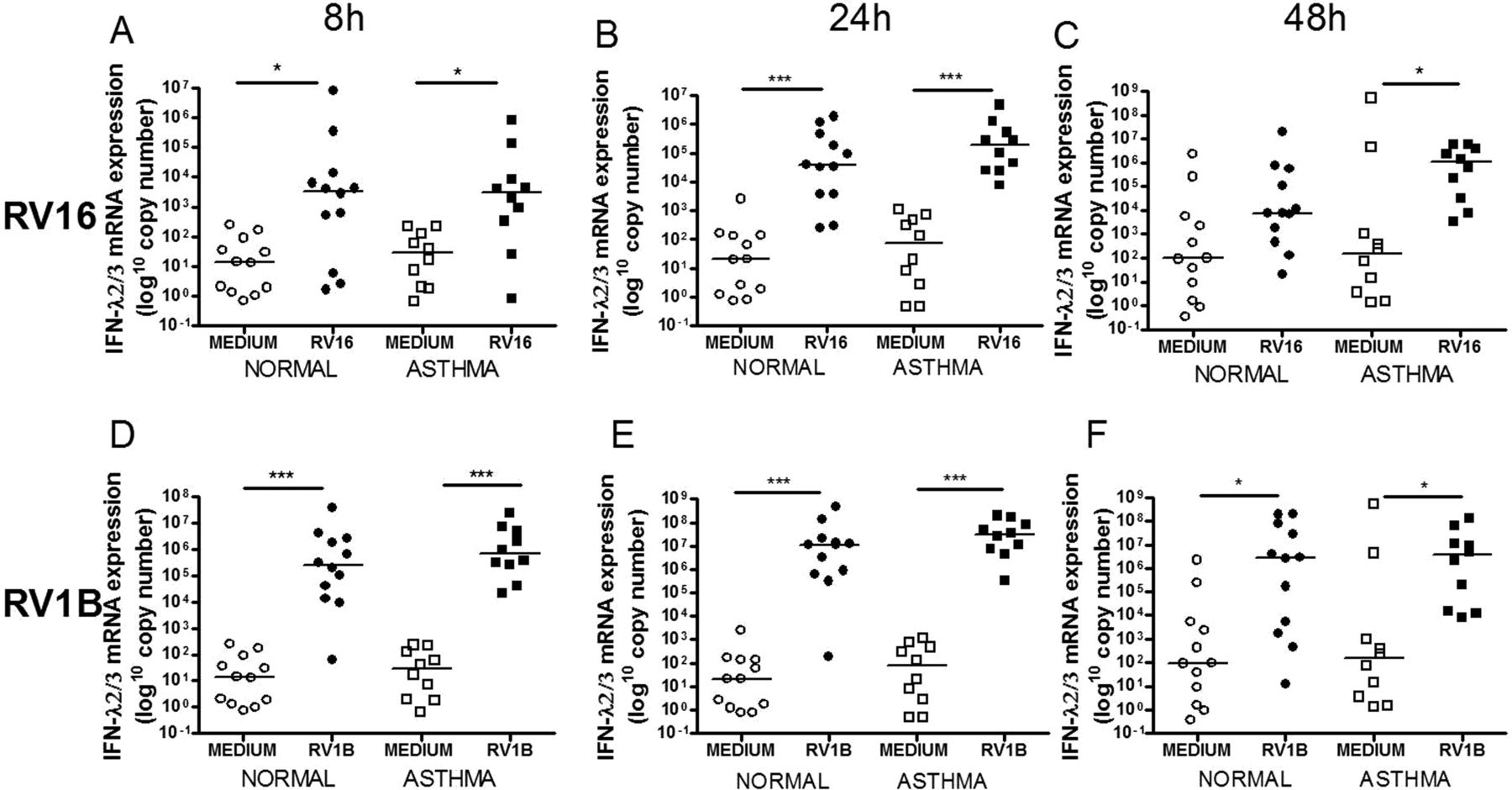
Interferon (IFN)-λ2/3 mRNA induction after rhinovirus (RV)-1B and RV16 was not different in subjects with asthma compared with healthy controls. IFN-λ2/3 mRNA at 8h (A), 24h (B) and 48h (C) post RV16 and 8h (D), 24h (E) and 48h (F) post RV1B infection. Squares denote subjects with asthma, circles denote healthy controls. *p<0.05, **p<0.01 and ***p<0.001.
RV-induced RV replication in HBECs was not significantly different between subjects with and without asthma
In addition to defective IFN induction, subjects with asthma have been observed to have increased RV replication and release.7 To assess whether this was present in the HBECs from subjects with asthma in this study, RV RNA copy number in cell lysates and live virus release into cell supernatants (measured by virus titration assay in TCID50/mL) were assessed.
Virus release increased over time following infection with RV16 and RV1B and levels were not significantly different between subjects with and without asthma. RV RNA levels were greatest at 48h for RV16 (figure 4A,C) and more variable for RV1B (figure 4B,D) and there were no significant differences between subject groups for RV RNA production with either virus at any time point.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rhinovirus (RV) release and viral RNA expression following RV16 and RV1B infection in human bronchial epithelial cells (HBECs) was not significantly different in subjects with asthma compared with healthy controls. Graphs denote live virus release into cell supernatant measured as tissue culture infectious dose 50 (TCID50)/mL for RV16 (A) and RV1B (B) and RV RNA copy numbers post RV16 (C) and RV1B (D). Error bars denote SDs. Squares denote subjects with asthma, circles denote healthy controls. No statistical significance was observed.
Discussion
This study has shown that the major IFN subtype induced in primary HBECs from subjects with asthma and healthy controls is IFN-λ, with IFN-λ1 and IFN-λ2/3 mRNAs being induced early at 8h for both RV serotypes and sustained induction being observed to 48h, while early and sustained induction of IFN-β was only observed for RV1B. IFN-λ protein levels induced by RV1B at 48h were substantially greater than levels of induction of IFN-β (p<0.001). No induction of IFN-α mRNA or protein was observed with either virus type. The importance of IFN-λ induction by HBECs in response to RV infection has been suggested previously by limited investigations in bronchial epithelial cells obtained commercially from three donors, and frozen for long periods of time, which identified significant induction of IFN-β and IFN-λ1 and IFN-λ2/3 mRNA at 24h by both RVs; however, this study failed to detect any IFN protein production.4 Our findings in primary HBECs cultured directly from the airways of 22 human subjects with and without asthma, therefore, provide much more rigorous confirmation that IFN-λ and, to a lesser degree, IFN-β are the major IFN subtypes induced by RV infection of primary HBECs.
IFN-β and IFN-λ protein were induced by RV1B but not RV16 infection in HBECs from subjects with asthma and healthy controls. The lack of induction following RV16 infection contrasts with the initial report of defective RV-induced IFN-β production, with a robust IFN protein response observed in subjects without asthma.7 Technical differences are not likely to be responsible for the differences in IFN protein induction observed between RV1B and RV16 in the present study as the viruses were cultured and stored in identical conditions, they had similar TCID50/mL in HeLa cells and they induced similar pro-inflammatory cytokine (IL-6 and IL-8) responses in HBECs and BEAS2B cells (data not shown). This study was also designed so that the sampling, cell culture techniques and viruses used replicated those used in the original report7 as closely as possible.
The IFN-β and IFN-λ protein levels measured in the present study were substantially lower compared with those observed in the initial reports of defective RV-induced IFN.5 ,7 IFN protein has been observed to vary with different RV subtypes and much lower levels of RV16-induced IFN-β protein have been reported6 ,19 since the original report.7 Other investigators have failed to detect RV16-induced IFN-β protein production despite significant mRNA induction,4 ,22 suggesting that bronchial epithelial cells may vary in their response to RV infection.
Our investigations in these new groups of subjects with atopic asthma and healthy controls failed to find any evidence for deficient type I or III IFN induction in primary HBECs from those with asthma following RV infection. The reason for the failure of this study to detect deficient IFN induction in HBECs from subjects with asthma is unclear as this was observed despite using similar experimental techniques and using the two RV serotypes previously shown to induce deficient responses.5–7 Major problems were experienced initially with HBEC cultures, resulting in the loss of many of the samples obtained (see online supplement for an explanation of these problems). Samples obtained by fresh bronchial brushings may contain a mixture of cell types which are then cultured in specialised bronchial epithelial cell culture medium for two passages (approx 30 days), which is likely to select out any other that may have been present. We have recently reported that type I IFN induction by exactly the same RV stocks used in the present study was deficient in BAL cells obtained from the larger group of the same subjects with asthma who provided HBECs for the present study, but of which only about half were successfully cultured compared with BAL cells from the larger group of these same normal subjects.8 It is therefore possible that there may be important differences between the participants with successful and unsuccessful HBEC cultures. Although all the participants had relatively mild asthma, a comparison of clinical characteristics raises the possibility that participants with unsuccessful cultures could have had less well controlled asthma than those with successful cultures (table 2). Participants with successful cultures had good asthma control according to Asthma Control Questionnaire (ACQ) scores—with an ACQ score of 0.5, with good asthma control being defined as an ACQ score of <0.75.23 In contrast, subjects with unsuccessful cultures had less well controlled asthma, with an ACQ score of 1.0. Participants with unsuccessful cultures had also been prescribed significantly greater numbers of courses of oral corticosteroids in the last year (p=0.007) and had a greater number of exacerbations. This could suggest that those with unsuccessful HBEC cultures could have had less well controlled asthma or were more prone to exacerbation than those with successful HBEC cultures.
Differences in clinical characteristics between subjects with asthma with successful and unsuccessful human bronchial epithelial cell (HBEC) cultures
A link between defective RV-induced type I IFN production and markers of more severe asthma has recently been identified, with the deficient type I IFN induction by RV in BAL cells from the same subjects studied in this report being associated with greater airway hyperresponsiveness and greater skin prick test positivity.8 This suggests that defective RV-induced IFN production may be a marker of more severe disease. Influenza virus induced IFN-α production by plasmacytoid dendritic cells has also recently been observed to correlate inversely with serum IgE levels,12 as has induction of the IFN-induced gene IP-10 following toll-like receptor 7 stimulation in PBMCs from subjects with asthma.24 This supports a link between severity of allergy and IFN production. A potential explanation for the lack of observation of defective RV-induced IFN production in this group of subjects with asthma is that defective IFN induction is a feature of less well controlled disease and that the subjects with asthma who had successful cultures in this study had asthma that was too well controlled for defective IFN to be detected. This may also explain why some other investigators have failed to identify IFN deficiency.14 ,15
Available information on disease severity from other reports of IFN responses in asthma is limited, and importantly, there are no data available on asthma control. In the genome-wide transcriptional analysis study, which did not report IFN deficiency in asthma, the nine study participants had a mean FEV1 % predicted of 80% and a PC20 methacholine of 1.65 mg/mL,15 which is similar to the patient group reported in this study (table 1). The six subjects with asthma recruited by Lopez-Sousa et al, who also did not report IFN deficiency in asthma,14 had potentially more severe disease (mean FEV1 % predicted 70% and PC20 methacholine 0.5 mg/mL). However, IFN-β was only measured in four of these subjects who were unspecified, making any further interpretation difficult. The original report of deficient RV-induced IFN production included subjects with asthma who were steroid treated or steroid naïve but did not give details regarding the level of asthma control.7 When the experiments in the study reported herein were conceived (2007), there was very limited literature available regarding defective IFN production in asthma and the potential link between asthma control and IFN responses had not been identified. Further investigation of RV-induced IFN induction in adults with well controlled and less well controlled asthma is therefore warranted. This is supported by our finding of defective RV-induced IFN in HBECs from children with severe asthma, using the same techniques and viruses as this study.25
Another potential explanation for the lack of defective RV-induced IFN production in this group of subjects with asthma is that defective IFN induction may not be related specifically to asthma, but to airway inflammation, and that the subjects with asthma in the different studies may have had differing degrees of airway inflammation. This is supported by the finding that defective RV-induced IFN production is also present in the other inflammatory airway diseases COPD18 and cystic fibrosis,19 diseases which are accompanied by much more severe airway inflammation than that observed in most subjects with asthma. Given that the subjects with asthma in this study whose HBECs were able to be studied had well controlled, mild asthma, it is likely that their airway inflammation was also mild.
In summary, this study has shown the major IFN subtype induced by RV in primary HBECs from subjects with asthma and healthy controls is IFN-λ, with IFN-β induced to a lesser degree, but IFN-α was not induced at all. The study failed to replicate the finding of deficient type I or III IFN in primary HBECs from subjects with asthma despite using similar techniques and identical viruses. The reason for this is unknown but it is possible that defective IFN induction may be related to the severity of asthma and allergy and that the group of subjects with asthma whose HBECs grew successfully had asthma that was too mild for a difference to be detected.
Acknowledgments
Joseph Eliahoo, statistical consultant at the statistical advisory service, Imperial College is acknowledged for statistical advice.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors Conceived and designed the experiments: SLJ, AS, MRE and MM. Subject recruitment and clinical data collections: AS, AdR and JM. Performed experiments: AS, VG and JH. Analysed the data: AS, MRE, SLJ and MM. Contributed to reagents/materials/analysis tools: OMK and MM. Wrote the paper: AS and SLJ.
-
Funding This work was supported by grants from the Medical Research Council (MRC project grant G0601236 and Centre Grant 100758, and a MRC Clinical Research Fellowship G0700552 (to AS)), the British Lung Foundation (grant P06/3), the Wellcome Trust (grant 083567/Z/07/Z for the Centre for Respiratory Infection), Asthma UK (grants 05/067 and 08/048, Asthma UK Fellowship RF07/04 (to MRE) and Asthma UK Clinical Chair CH11SJ (to SLJ)), Predicta FP7 Collaborative Project grant 260895, ERC FP7 advanced grant 233015 (to SLJ), AstraZeneca and the National Institute of Health Research Biomedical Research Centre and Clinical Lecturer funding schemes.
-
Competing interests SLJ is an author on patents relating to the use of IFNs in the treatment of exacerbations of airway disease and holds share options in Synairgen, a company developing IFNs for treatment of exacerbations of airway disease.
-
Ethics approval The study was approved by the St Mary's Hospital Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement All data from the study are in the process of being submitted for publication.
Linked Articles
- Airwaves
- PostScript