Article Text

Abstract
Background WNT signalling is activated during lung tissue damage and inflammation. We investigated whether lung epithelial expression of WNT ligands, receptors (frizzled; FZD) or target genes is dysregulated on cigarette smoking and/or in chronic obstructive pulmonary disease (COPD).
Methods We studied this in human lung epithelial cell lines and primary bronchial epithelial cells (PBEC) from COPD patients and control (non-)smokers, at baseline and on cigarette smoke extract (CSE) exposure.
Results CSE significantly decreased WNT-4, WNT-10B and FZD2 and increased WNT-5B mRNA expression in 16HBE, but did not affect WNT-4 protein. The mRNA expression of WNT-4, but not other WNT ligands, was lower in PBEC from smokers than non-smokers and downregulated by CSE in PBEC from all groups, yet higher in PBEC from COPD patients than control smokers. Moreover, PBEC from COPD patients displayed higher WNT-4 protein expression than both smokers and non-smokers. Exogenously added WNT-4 significantly increased CXCL8/IL-8, IL-6, CCL5/RANTES, CCL2/MCP-1 and vascular endothelial growth factor (VEGF) secretion in 16HBE, but did not affect the canonical WNT target genes MMP-2, MMP-9, fibronectin, β-catenin, Dickkopf and axin-2, and induced activation of the non-canonical signalling molecule p38. Moreover, WNT-4 potentiated the CSE-induced upregulation of IL-8 and VEGF.
Conclusions WNT-4 mRNA and protein levels are higher in PBEC from COPD patients than control (non-)smokers, while cigarette smoke downregulates airway epithelial WNT-4 mRNA, but not protein expression. As WNT-4 further increases CSE-induced pro-inflammatory cytokine release in bronchial epithelium, we propose that higher epithelial WNT-4 levels in combination with cigarette smoking may have important implications for the development of airway inflammation in COPD.
- Airway Epithelium
- COPD - Mechanisms
- COPD Pathology
Statistics from Altmetric.com
Key messages
What is the key question?
-
The main question of our study is whether dysregulated expression of WNT genes in airway epithelium contributes to the development of airway inflammation and remodelling on cigarette smoke exposure in COPD.
What is the bottom line?
-
We show for the first time that WNT-4 expression is higher in airway epithelium from COPD patients than from controls.
Why read on?
-
Our data further demonstrate that this may have important implications for airway inflammation in COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is a life-threatening disease with a worldwide increase in morbidity and mortality, characterised by irreversible airway obstruction and accelerated lung function decline.1 Cigarette smoking is the major risk factor for COPD. It can cause airway inflammation and abnormal tissue repair, important pathophysiological features of COPD that contribute to progressive lung function decline by inducing alveolar destruction and/or airway wall thickening and fibrosis. Airway epithelium is the first line of defence against environmental insults, such as inhaled cigarette smoke, and is an important source of pro-inflammatory mediators, including the neutrophil chemoattractant IL-8/CXCL8. Currently, it is still unclear how different outcomes of COPD develop from the same exposure to cigarette smoke.
The WNT signalling pathway plays an important role in lung development in utero. Recent data indicate that this pathway can be re-activated on tissue damage and inflammation in multiple organ systems, including the lungs.2 ,3 The WNT pathway consists of a complex signalling network. Autocrine or paracrine secreted WNT ligands activate frizzled receptors (FZD) that bind to co-receptors (LRP) and subsequently induce various downstream signalling pathways in the cell. Canonical WNT signalling results in β-catenin-mediated transcription of WNT target genes involved in tissue repair and remodelling,4 including growth factors (eg, vascular endothelial growth factor; VEGF), extracellular matrix (ECM) proteins (eg, fibronectin), matrix metalloproteinases (MMP) and cytokines (eg, IL-8). In the absence of WNT signalling, β-catenin is constitutively phosphorylated by its interaction with glycogen synthase kinase (GSK)-3β in the so-called β-catenin destruction complex and subsequently degraded. In the presence of active WNT ligands, the destruction complex is disrupted and β-catenin translocates to the nucleus to induce gene transcription. In addition, active WNT ligands can induce gene transcription through non-canonical signalling, including the p38, Ca2+/nuclear factor of activated T cell and RhoA-dependent pathways.5 ,6
Of interest, WNT signalling has been implicated in lung epithelial injury and repair processes.3 ,7 Furthermore, dysregulated expression of WNT genes and regulators has been observed in COPD fibroblasts,8 lung biopsies of COPD patients with emphysema9 and in airway epithelium of COPD patients10 compared to healthy controls. However, the role of WNT signalling in the development and progression of COPD is at present largely unknown.
In the current study we aimed to determine whether dysregulated expression of WNT genes on cigarette smoke exposure in airway epithelium may contribute to the development of airway inflammation and remodelling in COPD.
Methods
Epithelial cell culture
The human bronchial epithelial cell line 16HBE was kindly provided by Dr DC Gruenert (University of California, San Francisco, USA) and cultured on collagen-coated flasks in essential modified Eagle's minimum essential medium (Biowhittaker, Verviers, Belgium) containing 10% fetal calf serum.11 Primary bronchial epithelial cells (PBEC) were obtained from 13 current and ex-smoking COPD patients with global initiative for chronic obstructive lung disease stages II–IV1 by bronchial brushings according to standard guidelines.12 PBEC from controls were derived from Lonza (Walkersville, Maryland, USA; six smokers, five non-smokers) or from bronchial brushings in our own institute (seven smokers, six non-smokers).13 Age did not significantly differ between the three subject groups. See table 1 for subject characteristics. The medical ethics committee of our centre approved the study. Signed informed consent was obtained from the participants. PBEC were cultured in hormonally supplemented bronchial epithelium growth medium (Lonza) in collagen/fibronectin-coated flasks as described previously.14 PBEC were stored in liquid nitrogen before use in passage 3. For experimentation, cells were grown in duplicate in 24-well plates until 80–90% confluency, serum-deprived (16HBE) or placed on bronchial epithelial cell basal medium (Lonza) supplemented with transferrin and insulin (PBEC).
Characteristics of the subjects
Preparation of CSE
Cigarette smoke extract (CSE) was prepared as described previously.15 In short, Kentucky 3R4F research-reference cigarettes (The Tobacco Research Institute, Lexington, Kentucky, USA) were used without filter. Smoke from two cigarettes was bubbled through 25 ml medium (100% CSE). The extract was freshly prepared.
Stimulation of the cells
Cells were pretreated with or without p38 inhibitor SB203580 (1 μM; Tocris Bioscience, Bristol, UK) for 60 min. Subsequently, cells were exposed to 1–15% CSE and/or recombinant human WNT-4 (50 or 500 ng/ml; R&D systems) for 6 or 24 h and harvested in TRIreagent (MRC Inc.) for RNA isolation, in Laemmli buffer for cell lysate preparation or supernatants were collected. Within this range, CSE does not affect cell viability.16 See supplementary data (available online only) for the methods of RNA isolation, cyclic DNA synthesis, real-time PCR, immunodetection and cytokine measurements in supernatant.
siRNA downregulation of WNT-4
WNT-4 gene expression was downregulated in the 16HBE cells by the use of small interfering RNA as described before.11 In short, cells were seeded at 5×104 cells/well, transfected with WNT-4-targeted siRNA or non-targeting control oligonucleotides in a final concentration of 20 μM using lipofectamine 2000 and cultured for 3 days.
Statistical analysis
Data were analysed using the non-parametric rank-sum Mann–Whitney U test for analysis between subject groups and the non-parametric Wilcoxon signed rank test for paired observations within subject groups and for the 16HBE experiments (gene expression) or the Student's t test for analysis of the cytokine levels in 16HBE experiments, based on normal distribution.
Results
Expression of WNT ligands, WNT receptors and WNT target genes on CSE exposure in lung epithelial cell lines
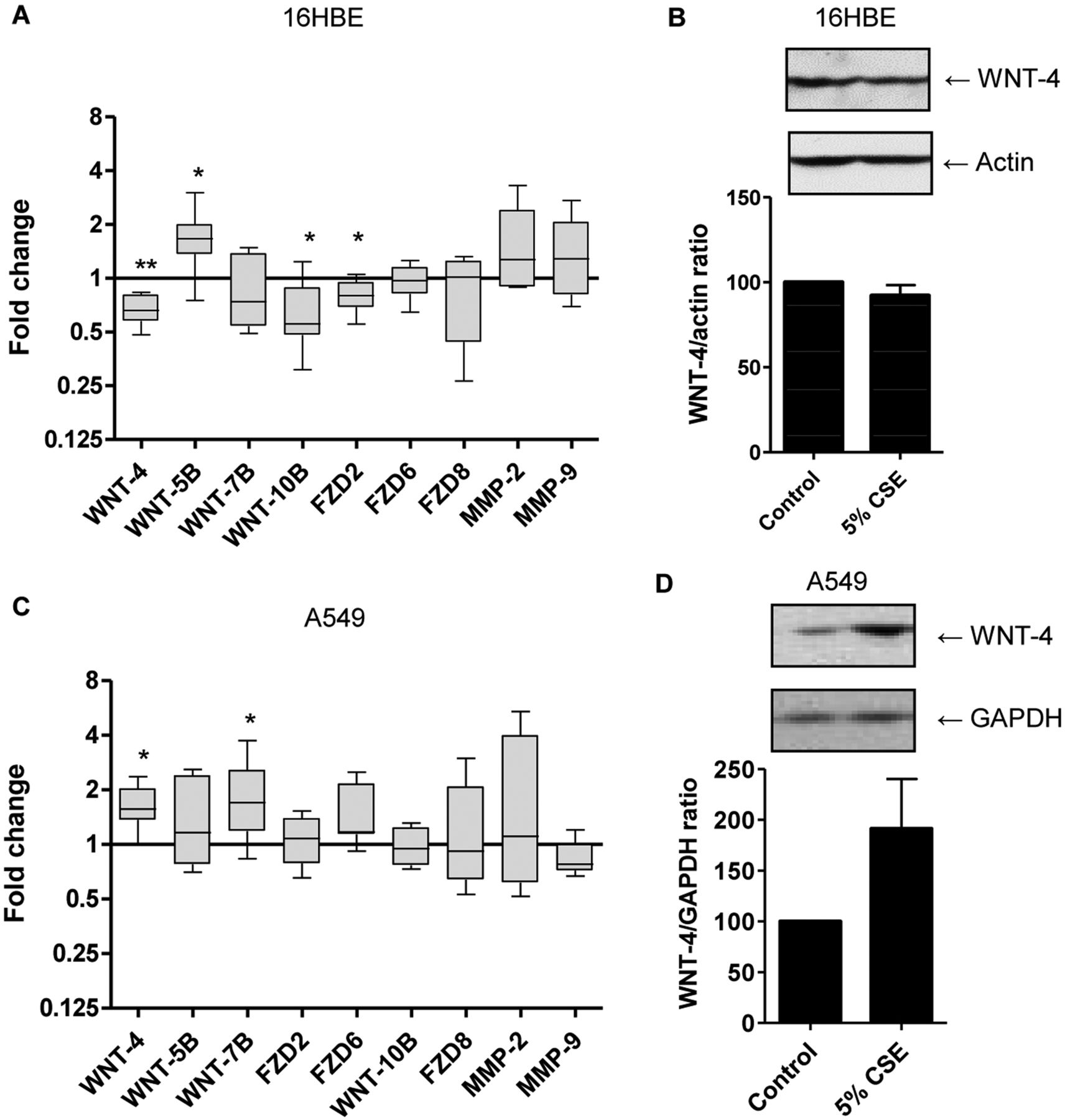
We first determined the expression of WNT ligands, receptors and target genes and their regulation by CSE in human bronchial epithelial 16HBE cells. We included heme oxygenase 1 as a positive control for the effects of CSE, showing a dramatic increase in 16HBE as well as in PBEC on treatment with 5% CSE (see supplementary figure E1A, available online only). We were unable to detect WNT-1, WNT-2, WNT-3A, WNT-5A, WNT-6, WNT-8B, WNT-11, WNT-16 and FZD1 messenger RNA expression in the 16HBE cells at baseline. Of the detected genes, the expression of WNT-7B, FZD6, FZD8 and the WNT target genes MMP-2 and MMP-9 remained unaltered on exposure to CSE for 6 h (figure 1A). Interestingly, 6 h CSE exposure downregulated the expression level of WNT-4, WNT-10B and FZD2 and upregulated WNT-5B mRNA expression in 16HBE cells, which could already be observed at 1% CSE (data not shown) and was significant for 5% and 10% CSE (figure 1A and see supplementary figure E1B, available online only). However, the downregulatory effect of CSE on WNT-4 mRNA could not be confirmed at the protein level (figure 1B).

Effects of cigarette smoke exposure on the expression of WNT genes and target genes in bronchial epithelial 16HBE cells and alveolar epithelial A549 cells. (A) 16HBE cells were serum-deprived overnight, incubated with or without 5% cigarette smoke extract (CSE) for 6 h and RNA was isolated. WNT gene (WNT-4, WNT-5B, WNT-7B, WNT-10B and FZD2, FZD-6, FZD-8) and target gene (MMP-2, MMP-9) expression was related to the expression of the housekeeping genes β2μG and PPIA. mRNA levels are expressed as fold change compared to the unstimulated control value (2−ΔΔCt, n=6–9) and medians are indicated. (B) 16HBE cells were exposed to 5% CSE for 24 h. Total cell lysates were prepared and WNT-4 was detected by western blotting. Actin was used as loading control. Densitometry was performed and levels were related to actin levels. A representative blot and the WNT-4/actin ratios (mean±SEM) are depicted (n=5). (C) A549 cells were serum-deprived overnight, incubated with or without 5% CSE for 6 h and RNA was isolated. WNT gene (WNT-4, WNT-5B, WNT-7B, WNT-10B and FZD2, FZD-6, FZD-8) and target gene (MMP-2, MMP-9) expression was related to the expression of the housekeeping genes β2μG and PPIA. mRNA levels are expressed as fold change compared to the unstimulated control value (2−ΔΔCt, n=6–9) and medians are indicated. (D) A549 cells were exposed to 5% CSE for 24 h. Total cell lysates were prepared and WNT-4 was detected by western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. Densitometry was performed and levels were related to GAPDH levels. A representative blot and the WNT-4/GAPDH ratios (mean±SEM) are depicted (n=3). *p<0.05 and **p<0.01 between the indicated values.
In contrast to 16HBE cells, 5–10% of CSE did not decrease but instead increased the expression level of WNT-4 and FZD2 as well as WNT-7B in the alveolar cell line A549 (figure 1C and see supplementary figure E1C, available online only). The upregulatory effect of 5% CSE on WNT-4 mRNA expression in A549 cells could be confirmed at the protein level (figure 1D).
Expression of WNT ligands and receptors in bronchial epithelial cells from COPD patients and smoking and non-smoking controls
We next assessed whether the differences in WNT gene expression on CSE exposure in the bronchial epithelial cell line were reflected in PBEC from COPD patients (current and ex-smokers), control smokers and non-smokers. We observed a similar WNT gene expression profile as in the 16HBE cells (see figure 2A), although PBEC expressed detectable levels of WNT-5A mRNA, while we were not able to detect WNT-10B and FZD6 mRNA in these cells. In line with the in-vitro effects of CSE, we observed lower expression of WNT-4 mRNA, but none of the other WNT genes, in PBEC from control smokers compared to non-smokers (figure 2A). Importantly, WNT-4 expression levels in PBEC from COPD patients were significantly higher than in the control smokers, while they were not significantly different from the non-smokers. WNT-4 mRNA levels did not differ between current and ex-smokers within the COPD group (see supplementary figure E2A, available online only) nor did they differ between normal human bronchial epithelium and brushed PBEC (see supplementary figure E2B, available online only). Furthermore, we studied the effect of CSE on WNT-4 mRNA expression, demonstrating that 5% CSE exposure (6 h) reduced WNT-4 expression in PBEC from all subject groups, similar to the effects observed in 16HBE (figure 2B). We aimed to validate the differences in WNT-4 expression between the groups at the protein level by immunodetection and observed that COPD epithelium displayed higher WNT-4 levels than both control smokers and non-smokers (figure 2C). In line with the findings in 16HBE, the lower WNT-4 mRNA levels in PBEC from smoking compared to non-smoking controls were not accompanied by lower protein levels. On the contrary, higher WNT-4 mRNA expression in PBEC from COPD patients compared to control smokers corresponded to increased protein levels of WNT-4. Of interest, WNT-4 protein levels in PBEC from COPD patients were also higher compared to non-smokers.

mRNA expression of WNT-4, but none of the other WNT genes is higher in primary bronchial epithelial cell (PBEC) from chronic obstructive pulmonary disease (COPD) patients than from healthy control smokers. (A) Cells were growth-factor deprived overnight, cultured in fresh growth factor-free medium for 2 h and mRNA was isolated. We assessed WNT gene (WNT-4, WNT-5B, WNT-7B, WNT-10B and FZD2, FZD-6, FZD-8) expression in PBEC from stage II–IV COPD patients (ex-smokers n=8, current smokers n=3), control smokers (n=10, including n=5 from Lonza) and non-smokers (n=11, including n=5 from Lonza) at baseline. Gene expression was related to the expression of the housekeeping genes β2μG and PPIA. mRNA levels are expressed as fold change compared to one of the non-smoking controls (2−ΔΔCt) and medians are indicated. (B) In a subgroup of non-smokers (n=5), control smokers (n=6) and COPD patients (ex-smokers n=3, current smokers n=5) cells were growth-factor deprived overnight, cultured with or without cigarette smoke extract (CSE) for 6 h and mRNA was isolated. WNT-4 expression was related to the expression of the housekeeping genes β2μG and PPIA. mRNA levels on CSE exposure expressed as fold change compared to the corresponding unstimulated control value (2−ΔΔCt) and medians are indicated. (C) In a subgroup of non-smokers (n=7 including n=4 from Lonza), control smokers (n=6 including n=4 from Lonza) and COPD patients (ex-smokers, n=8), total cell lysates were prepared and WNT-4 was detected by western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. Densitometry was performed and levels were related to GAPDH levels. WNT-4/GAPDH ratios are depicted (n=7–8 per group) and a representative blot is shown. *p<0.05 and **p<0.01 between the indicated values.
WNT-4 induces expression of pro-inflammatory cytokines and VEGF, but not additional canonical target genes in 16HBE
Given the increased WNT-4 levels in COPD epithelium, we investigated the consequences of active WNT-4 signalling, both in the absence and presence of cigarette smoke. We first exposed 16HBE cells to recombinant human WNT-4 (50 and 500 ng/ml) for 24 h in the absence of CSE and studied effects on pro-inflammatory cytokines and remodelling parameters. Interestingly, WNT-4 significantly increased the release of WNT targets and pro-inflammatory mediators IL-8, IL-6, CCL2 and CCL5 (figure 3A). Similar effects of WNT-4 were observed on IL-1α, CCL3 and CCL4 release, but these effects failed to reach statistical significance, while levels of IL-1β did not increase on WNT-4 stimulation (data not shown). Furthermore, WNT-4 (50 and 500 ng/ml) increased the secretion of the growth factor VEGF over baseline levels, with a significant effect at 500 ng/ml (figure 3A). We also studied gene expression of additional targets of the WNT signalling pathway, including MMP-2 and MMP-9, fibronectin, β-catenin, DKK1, axin-2, WIF and WISP1. In contrast to the data on pro-inflammatory cytokines and VEGF, WNT-4 did not significantly alter MMP-2, MMP-9, fibronectin, β-catenin, DKK1 or axin-2 expression, while we were unable to detect WIF and WSP1 in the presence or absence of WNT-4 (figure 3B). In line with the lack of effect on these canonical WNT targets, WNT-4 did not induce transcriptional activity of the β-catenin-driven reporter construct TopFLASH (see supplementary figure E3, available online only). In accordance with a role for endogenous WNT-4 in the secretion of pro-inflammatory cytokines, the downregulation of WNT-4 by siRNA resulted in reduced IL-8 secretion compared to cells treated with non-targeting control siRNA (figure 3C). In line with the findings in 16HBE, WNT-4 augmented IL-8 secretion in A549 cells (see supplementary figure E4, available online only). To elucidate whether WNT-4 affects non-canonical pathways or the activity of GSK-3β in 16HBE cells, we studied the phosphorylation of p38, RhoA downstream molecule myosin light chain and GSK-3β. WNT-4 did not induce phosphorylation of myosin light chain or GSK-3β (see supplementary figure E5, available online only), but substantially increased the levels of phosphorylated of p38 (figure 3D). In line with a role for p38 in the WNT-4-induced effects in 16HBE, we observed that the p38 inhibitor SB203580 abrogated the WNT-4-induced upregulation of IL-6 (figure 3E), the cytokine that showed the most pronounced increase on WNT-4 stimulation.

Recombinant human WNT-4 increases the release of pro-inflammatory cytokines/chemokines IL-8, IL-6, CCL2 and CCL5 and growth factor vascular endothelial growth factor (VEGF) but does not affect the expression of various targets of the canonical WNT signalling pathway and induces non-canonical signalling in 16HBE cells. 16HBE cells were serum-deprived overnight and treated with or without 50 and 500 ng/ml rhWNT-4, in the presence and absence of p38 inhibitor SB203580. Total cell lysates were prepared on 0–240 min, mRNA was isolated and cytokines levels were assessed in cell-free supernatants on 24 h of WNT-4 exposure. (A) IL-8, IL-6, CCL2 and CCL5 levels were measured by a multiplex ELISA and VEGF levels were measured by a conventional ELISA. Cytokine levels are expressed as a percentage of the unstimulated control or as absolute values (mean±SEM, n=3). #p<0.05 versus the unstimulated control. (B) mRNA expression (MMP-2, MMP-9, fibronectin, β-catenin, DKK1 and axin-2) was related to the expression of the housekeeping genes (n=4). mRNA levels are expressed as fold change compared to the unstimulated control (2−ΔΔCt). (C) 48 h before to serum starvation, cells were transfected with non-targeting control siRNA (siRNA con) or siRNA specific for WNT-4 (siRNA WNT-4). mRNA was isolated and cell-free supernatants were harvested after 24 h. WNT-4 mRNA expression was related to the expression of the housekeeping genes β2μG and PPIA (n=4). mRNA levels on WNT-4 siRNA treatment are expressed as fold change compared to the corresponding siRNA control (2−ΔΔCt). IL-8 levels were measured in cell-free supernatants by conventional ELISA and expressed as a percentage of the siRNA control (mean±SEM, n=4) *p<0.05. (D) phospho-p38 was detected by western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as loading control. Densitometry was performed and levels were related to GAPDH levels. A representative of three independent experiments is shown and the corresponding diagram refers to the ratios and means (±SEM, n=3). (E) IL-6 levels were measured in cell-free supernatants by conventional ELISA and as absolute values (mean±SEM, n=3). *p<0.05 between the indicated values.
WNT-4 augments the CSE-induced upregulation of IL-8 and VEGF in 16HBE cells
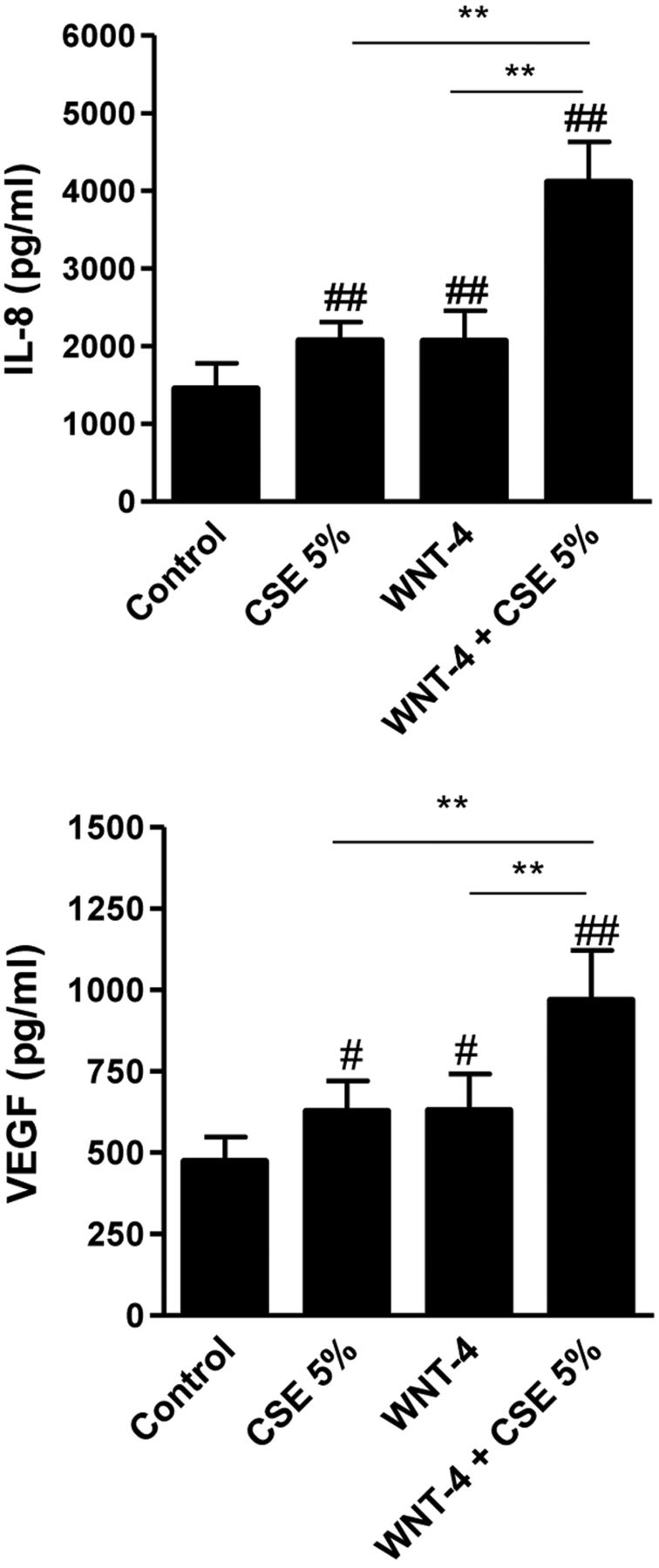
Finally, we tested whether WNT-4 is able to modulate the effect of CSE on IL-8 and VEGF. Five per cent of CSE induced a modest, but significant, increase in the release of both VEGF and IL-8 in 16HBE cells (figure 4). As demonstrated above, WNT-4 also increased VEGF and IL-8 release. Of interest, the presence of WNT-4 potentiated the upregulatory effect of 5% CSE on VEGF as well as IL-8 secretion, increasing the fold-induction from 128±4% to 196±19% and from 156±18% to 272±23%, respectively (figure 4). Exposure of airway epithelium to CSE in the presence of high levels of WNT-4 may thus lead to increased pro-inflammatory epithelial responses.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Recombinant human WNT-4 augments the cigarette smoke extract (CSE)-induced release of IL-8 and vascular endothelial growth factor (VEGF). 16HBE cells were serum-deprived overnight and treated with or without 5% CSE, in the presence and absence of 500 ng/ml rhWNT-4. Cytokine levels were assessed on 24 h of WNT-4 exposure in cell-free supernatants by ELISA. Absolute cytokine levels are shown (mean±SEM, n=6). **p<0.01 between the indicated values. #p<0.05 and ##p<0.01 versus the unstimulated control.
Discussion
The WNT pathway is activated during lung inflammatory and remodelling processes and plays a key role in epithelial repair responses. Our current data indicate a role for aberrant regulation of the WNT pathway, especially WNT-4, in airway inflammation in COPD. We show that WNT-4 mRNA and protein expression is higher in epithelial cells from COPD patients than control (non-)smokers. This may have important consequences, because exogenously added WNT-4 increased the release of IL-8, IL-6, CCL2, CCL5 and VEGF and additionally augmented the CSE-induced release of IL-8 and VEGF in bronchial epithelial cells. Therefore, high levels of WNT-4 may amplify and sustain pro-inflammatory responses, especially when epithelial cells are exposed to cigarette smoke.
To our knowledge, we are the first to demonstrate the higher expression of WNT-4 in epithelium from COPD patients compared to age-matched control smokers. WNT-4 mRNA levels were increased compared to the control smokers only, whereas WNT-4 protein levels were increased compared to both control smokers and non-smokers. Our studies were performed in cultured epithelial cells, and we found no differences between current and ex-smoking COPD patients, indicating that these changes are persistent and irrespective of smoking status, possibly resulting from (epi)genetic differences in WNT-4 or regulators of WNT-4. Cigarette smoke exposure reduced WNT-4 mRNA (but not protein) expression in 16HBE cells and PBEC, while PBEC from control smokers displayed reduced levels of WNT-4 mRNA (but not protein) compared to non-smokers, further suggesting that the increased WNT-4 expression in COPD epithelium is not due to smoking per se. The discrepancy between WNT-4 mRNA and protein levels indicates that WNT-4 expression is also regulated at the post-translational level, for instance by the downregulation of microRNA that can suppress gene expression at the translational level. At the same time, transcriptional repression of WNT-4 mRNA may occur, for example, by the antioxidant p21Waf'1/CIP1 that is specifically known to suppress WNT-4 expression.17 An imbalance in such parallel regulatory processes on repeated exposure to cigarette smoke could contribute to increased airway epithelial WNT-4 expression in COPD patients.
In contrast to our current findings, Wang et al10 reported that epithelial expression of WNT pathway genes (FZD4, FZD6 and FZD8) and target genes (eg, axin-1, VEGF) was reduced in both control smokers and COPD patients when compared to non-smokers, which was accompanied by an increase in the WNT pathway inhibitor secreted-frizzled related protein (SFRP)2. The authors suggested that the upregulation of SFRP2 may be responsible for the downregulation of the WNT pathway, allowing for epithelial recovery and differentiation,10 because SFRP2 was specifically expressed in the ciliated epithelial cell population. We were not able to detect SFRP2 mRNA in the epithelial cell lines or primary cultures or in primary cells grown at the air–liquid interface, allowing for mucociliary differentiation. Dissimilarities between the studies may be explained by the fact that we used bronchial cells from the second to fifth branch that were cultured for 2–3 weeks before experimentation, whereas Wang and co-workers10 used small airway epithelial cells from the 10th to 12th branch that were subjected to microarray analysis directly on collection. Thus, differences in the regulation of the WNT pathway could be due to different factors present in the epithelial microenvironment. In addition to the study of Wang and co-workers,10 Kneidinger et al9 observed higher levels of WNT-10B, but not WNT-2, WNT-3A, WNT-7B, FZD1 and FZD4, in parenchymal lung tissue specimens of COPD stage IV patients. In line with our findings in 16HBE, Kneidinger and co-workers9 found reduced WNT-10B mRNA expression levels in lung tissue of mice exposed to cigarette smoke. Furthermore, they observed that the expression of WNT-10B and additional WNT (target) genes, including WNT2, FZD1 and axin1/2, was downregulated in an elastase-induced emphysema mouse model, in which WNT/β-catenin activation exhibited beneficial effects.9 These studies point towards a protective rather than a detrimental role of the WNT pathway in lung emphysema. Potentially, activation of the WNT pathway plays an opposite role in the airways. Cigarette smoking can induce the loss of ECM in lung parenchyma and increased ECM deposition in the airway walls. We hypothesise that cigarette smoke differentially affects the WNT pathway in lung parenchyma and the airways of COPD patients, similar to the dual role of VEGF in the alveolar and bronchial compartment.18 In this respect, reduced VEGF activity causes alveolar epithelial cell apoptosis in emphysema, whereas increased sputum levels of VEGF have been associated with airflow limitation in patients with bronchitis.18 ,19 We observed that CSE increases mRNA and protein expression of WNT-4 in alveolar A549 cells, while it decreases WNT-4 mRNA expression in bronchial epithelial cells. Because of technical limitations, we did not confirm these data in primary alveolar epithelial cells, and we cannot exclude the possibility that the differences are due to altered sensitivity of carcinoma A549 cells to cigarette smoke, for example, with respect to antioxidant capacity.
Given the upregulatory effects of WNT-4 on the release of VEGF and pro-inflammatory cytokine/chemokines, we propose that increased WNT-4 signalling contributes to airway remodelling and airway inflammation in COPD. IL-8 is a well-known chemoattractant of neutrophils. CCL2 and CCL5 attract macrophages and eosinophils, respectively, and levels of these chemokines as well as IL-6 are known to be increased in COPD airways.20 ,21 Furthermore, VEGF has been implicated in vascular airway remodelling in COPD patients with bronchitis.18 ,19 WNT-4 did not induce other remodelling parameters and classic WNT target genes, for example, MMP-2, MMP-9, β-catenin, DKK1 and axin-2 in our setting. Our data further suggest that WNT-4 does not activate canonical WNT signalling in bronchial epithelium. Instead, WNT-4 appeared to activate the non-canonical p38 pathway in 16HBE cells. In line with our results on the involvement of p38 in the WNT-4-induced effects, p38 activation has previously been described to induce VEGF release in airway smooth muscle cells22 and is well known to be involved in pro-inflammatory gene expression. Accordingly, WNT-4 is usually described as an activator of non-canonical WNT signalling, ie, through FZD6 activation.23 Our study showed that cigarette smoke reduces FZD2 expression in 16HBE cells along with the downregulatory effect on WNT-4, suggesting a link between WNT-4 and FZD2 signalling. However, we are unaware of data supporting a role for WNT-4 as FZD2 ligand, and we did not observe this link in primary bronchial epithelium from our subjects.
Together, our data show that WNT-4 expression is higher in bronchial epithelial cells from COPD patients than from age-matched control smokers. Increased WNT-4 expression may have important consequences, because WNT-4 increased VEGF, IL-8, IL-6, CCL2 and CCL5 release from epithelial cells, and moreover, WNT-4 amplified the upregulatory effect of cigarette smoke on VEGF and IL-8 release. Future studies to reveal the mechanisms involved in the increased expression of WNT-4 in airway epithelium from COPD patients may provide insight into a novel therapeutic approach in the treatment of airway inflammation and remodelling in COPD.
Acknowledgments
The authors would like to thank L den Boef and U Brouwer for their help with the mouse model and M van der Toorn for his help with the multiplex ELISA.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Funding This study was supported by the Dutch Royal Academy of Sciences (KNAW).
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval This study was approved by the medical ethics committee of the University Medical Center, Groningen.
-
Provenance and peer review Not commissioned; internally peer reviewed.