Article Text

Abstract
Background Fibroblast to myofibroblast transition is believed to contribute to airway remodelling in lung diseases such as asthma and chronic obstructive pulmonary disease. This study examines the role of aclidinium, a new long-acting muscarinic antagonist, on human fibroblast to myofibroblast transition.
Methods Human bronchial fibroblasts were stimulated with carbachol (10−8 to 10−5 M) or transforming growth factor-β1 (TGF-β1; 2 ng/ml) in the presence or absence of aclidinium (10−9 to 10−7 M) or different drug modulators for 48 h. Characterisation of myofibroblasts was performed by analysis of collagen type I and α-smooth muscle actin (α-SMA) mRNA and protein expression as well as α-SMA microfilament immunofluorescence. ERK1/2 phosphorylation, RhoA-GTP and muscarinic receptors (M) 1, 2 and 3 protein expression were determined by western blot analysis and adenosine 3′-5′ cyclic monophosphate levels were determined by ELISA. Proliferation and migration of fibroblasts were also assessed.
Results Collagen type I and α-SMA mRNA and protein expression, as well as percentage α-SMA microfilament-positive cells, were upregulated in a similar way by carbachol and TGF-β1, and aclidinium reversed these effects. Carbachol-induced myofibroblast transition was mediated by an increase in ERK1/2 phosphorylation, RhoA-GTP activation and cyclic monophosphate downregulation as well as by the autocrine TGF-β1 release, which were effectively reduced by aclidinium. TGF-β1 activated the non-neuronal cholinergic system. Suppression of M1, M2 or M3 partially prevented carbachol- and TGF-β1-induced myofibroblast transition. Aclidinium dose-dependently reduced fibroblast proliferation and migration.
Conclusion Aclidinium inhibits human lung fibroblast to myofibrobast transition.
- Fibroblast
- myofibroblast
- anticholinergic
- asthma
- COPD
- airway epithelium
- interstitial fibrosis
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Key messages
What is the key question?
Does aclidinium, an anticholinergic agent, affect human fibroblast to myofibroblast transition?
What is the bottom line?
Aclidinium dose-dependently inhibits human lung fibroblast to myofibroblast transition induced by carbachol and TGF-β1 stimulation.
Why read on?
This is the first report of the mechanistic and functional involvement of a non-neuronal cholinergic system in the transition of human lung fibroblasts to myofibroblasts. We provide evidence that the use of anticholinergic agents such as aclidinium for the treatment of lung disease may play a role in regulatory fibrotic remodelling.
Introduction
Remodelling is a pathological feature observed in the airways of patients with asthma and chronic obstructive pulmonary disease (COPD), and the extent and progression of these structural alterations correlate with disease severity.1 2 Fibrotic alterations as part of these structural changes are also observed in both asthma and COPD, although their nature, localisation and extent are different. Under chronic inflammatory conditions, resident lung fibroblasts are activated and transformed into a more contractile, proliferative and secretory-active myofibroblast phenotype characterised by an increased expression of extracellular matrix components and α-smooth muscle actin (α-SMA), which contribute to the increase in lung remodelling progression and airway bronchoconstrictor responsiveness.3
Parasympathetic activity is increased in airway inflammation, which is the basis for the use of anticholinergic therapy in asthma and COPD.4 Anticholinergics constitute a particularly important bronchodilator therapy in COPD and certain forms of asthma.5 Furthermore, anticholinergics have shown potential anti-inflammatory and anti-remodelling effects in animal models,6 which may be of added value to their classical bronchodilator effects. Recent evidence indicates that acetylcholine production in the airways is not restricted to the parasympathetic nervous system but can also be released from non-neuronal origins.7 Dysfunction of the non-neuronal cholinergic system appears to be involved in the pathophysiology of asthma and COPD.8 In this regard, it has been suggested that fibroblasts are endowed with this system and could represent a previously unappreciated regulatory pathway in pulmonary remodelling.9 Muscarinic receptor stimulation has been shown to induce profound proliferation and collagen secretion of human lung fibroblasts.10 11 However, the role of the cholinergic system in fibroblast to myofibroblast transition, as well as the mechanisms that underlie fibroblast activation, remain unclear.
Aclidinium bromide (Almirall, Barcelona, Spain) is a novel long-acting muscarinic antagonist that has reached phase III clinical development for COPD treatment.12 In preclinical studies, aclidinium demonstrated potent muscarinic antagonist activity comparable to ipratropium and tiotropium, with a long duration of action.13 The aim of the present study was to investigate the effect of aclidinium on regulating carbachol-induced human lung fibroblast to myofibroblast transition as well as the mechanism involved in this process. In addition, since transforming growth factor-β1 (TGF-β1) is the main known factor responsible for myofibroblast formation and progression,14 we examined the effects of aclidinium on TGF-β1-induced fibroblast to myofibroblast transition and the possible link between cholinergic and TGF-β1 pathways.
Methods
Isolation and cultivation of human fibroblasts
Human lung fibroblasts were dissected from lobar or main bronchus tissue obtained from patients undergoing surgery for lung carcinoma, as previously described.10 Data presented throughout the study were from human bronchial fibroblasts obtained from patients who smoked. For comparison, bronchial fibroblasts were also isolated from patients with COPD and only used to explore the effect of aclidinium on carbachol- or TGF-β1-induced myofibroblast markers. Cultivation and characterisation of fibroblasts were performed as described elsewhere10 11 (for details and clinical data of patients see online supplement).
Stimulation of human fibroblasts
Before stimulation, subconfluent cell monolayers were deprived of serum for 24 h. Human bronchial fibroblasts were stimulated with carbachol (Sigma, Madrid, Spain: catalogue no. C4382) or human TGF-β1 (2 ng/ml; Sigma: catalogue no. T7039) for the indicated times, replacing culture medium and stimulus every 24 h. Aclidinium, human acetylcholinesterase (10 U/ml; AChE; Sigma: catalogue no. C1682), dibutyryl adenosine 3′-5′ cyclic monophosphate sodium salt (1 mM; dbcAMP; Sigma: catalogue no. D0260), PD98050 (10 μM; Sigma: catalogue no. P215), Y27632 (10 μM; Sigma: catalogue no. Y0503), pirenzepine (described as M1 antagonist, 1 μM; Sigma: catalogue no. P7412), methoctramine (described as M2 antagonist, 1 μM; Sigma: catalogue no. M105) and p-fluoro-hexahydro siladifenidol (pFHHSid) (described as M3 antagonist, 1 μM; Sigma: catalogue no. H127) were added 30 min before stimulus. Monoclonal anti-human TGF-β1 monoclonal antibody (4 μg/ml; anti-TGF-β1; R&D Systems, Madrid, Spain; catalogue no. AB-246-NA) was added 30 min before stimulus to block the culture supernatant active form of TGF-β1, as previously outlined.15
Real-time RT-PCR
Total RNA isolation and specific cDNA amplification for muscarinic acetylcholine receptors M1, M2, M3, α1(I)-collagen (col type I), α-SMA and TGF-β1 were referenced to glyceraldehyde phosphate dehydrogenase as an endogenous control and normalised to the control group, as previously described16 17 (see online supplement for details).
Transfection of siRNAs
Small interfering RNA (siRNA) experiments for M1, M2 and M3 were performed in lung fibroblasts as previously outlined16 17 using lipofectamine-2000 (Invitrogen, Paisley, UK) 2 μl/ml as transfection reagent (see online supplement for details).
Western blotting
Western blot analysis was used to detect changes in col type I (138 kD), α-SMA, TGF-β1 (40−60 kD), p-ERK1/2 (42−44 kD), RhoA-GTP (22 kD), M1 (52 kD), M2 (70 kD), M3 (75 kD) and choline acetyltransferase (ChAT; 65 kD) (see online supplement for details).
Immunofluorescence
Fibroblasts were fixed, permeabilised and immunostained with anti-α-SMA (1:200) antibody, followed by secondary anti-FITC, as previously outlined17 (see online supplement for details).
Enzyme-linked immunosorbent assays
Quantitative ELISAs for TGF-β1 and acetylcholine (ACh) were performed with supernatants of subconfluent human lung fibroblasts on a six-well plate following 48 h of stimulation with Quantikine human TGF-β1 immunoassay (R&D Systems; catalogue no. 891124) and ACh assay kit (Abcam, UK; catalogue no. ab65345), respectively.
The cell content of cAMP was measured as previously described.18 Cells were placed in Dulbecco's modified Eagle medium with 1% fetal calf serum (FCS) for 24 h before measurements to arrest growth. The experimental protocol consisted of incubation of cells with aclidinium for 30 min followed by addition of carbachol (10−5 M) for 10 min and isoprenaline (10−6 M) for another 10 min. These concentrations and times of incubation were selected from the literature.19 Total cAMP content was determined using a commercially available biotrack enzyme immunoassay kit (reference RPN2251; Amersham, Bucks, UK) (see online supplement for details).
Cell proliferation assay and cell migration
Human bronchial fibroblast proliferation and migration were measured by colorimetric immunoassay based on BrdU incorporation and wound closure assay, as previously outlined20 (for details see online supplement).
Analysis of results
Results are expressed as mean (SEM). Comparisons over time or across different stimulations on matched fibroblast samples of one subject for n independent subjects were analysed by one-way repeated measures analysis of variance (ANOVA) with 95% CIs. For separate comparisons of each stimulation, post hoc Bonferroni-Holm tests were performed. A p value <0.05 was considered statistically significant.
Results
Aclidinium inhibits carbachol- and TGF-β1-induced collagen type I and α-SMA expression in human lung fibroblasts
Carbachol (10−5 M) augmented collagen type I and α-SMA mRNA expression, reaching peak values at 48 h after carbachol exposure (figure 1A,B). Furthermore, carbachol dose-dependently increased collagen type I and α-SMA protein and mRNA expression reaching significant values at 10−5 M (figure 1C,D). We therefore selected this carbachol concentration for the rest of the experiments.
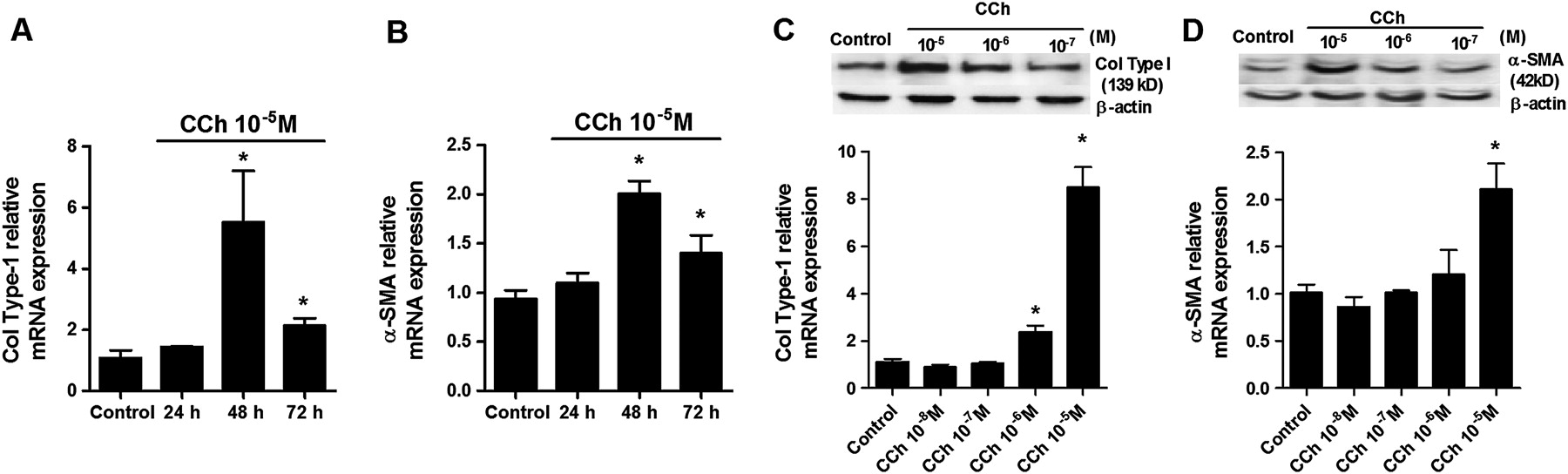
Carbachol (CCh) time- and dose-dependently increases collagen type I (col type I) and α-smooth muscle actin (α-SMA) in human lung fibroblasts. Human lung fibroblasts were stimulated with CCh at the indicated times (A, B) or for 48 h at the indicated concentrations (C, D). (A−D) After incubation, RNA was extracted and subjected to RT-PCR with col type I and α-SMA-specific primers and probes. (C, D) After incubation, total protein was extracted and western blot analysis was performed with specific antibodies for col type I and α-SMA. Each graph represents the mean of four individual experiments. One-way repeated measures analysis of variance (ANOVA): p<0.001 (A−D). *p<0.05 compared with solvent controls.
Aclidinium dose-dependently reduced carbachol-induced collagen type I and α-SMA protein and mRNA expression, reaching a maximal inhibitory value at 10−7 M (figure 2A,B; black bars) in bronchial fibroblasts from smokers. In other experiments, TGF-β1 caused a pronounced increase in collagen type I and α-SMA protein and mRNA expression which was almost completely blocked by aclidinium 10−7 M (figure 2C,D; black bars) in bronchial fibroblasts from smokers. For comparison, bronchial fibroblasts from patients with COPD were stimulated with carbachol or TGF-β1, reaching slightly higher levels of collagen type I and α-SMA mRNA transcripts than those observed in fibroblasts from smokers (figure 2A–D; white bars). Aclidinium dose-dependently reduced carbachol- and TGF-β1-induced collagen type I and α-SMA mRNA in a similar fashion to that in fibroblasts from smokers (figure 2A–D; white bars).

Carbachol (CCh) and transforming growth factor β1 (TGF-β1)-induced collagen type I (col type I) and α-smooth muscle actin (α-SMA) expression is inhibited by aclidinium (ACL). Human lung fibroblasts from smokers (black bars) or patients with chronic obstructive pulmonary disease (COPD) (white bars) were stimulated with CCh (A, B) or TGF-β1 (C, D) for 48 h. Aclidinium was added to the medium at the indicated concentrations 30 min before CCh (A, B) or TGF-β1 stimulation (C, D). Total protein and RNA were extracted after the incubation period. The top graphs represent values of densitometry of col type I (A, C) or α-SMA (B, D) protein expression relative to β-actin and normalised to solvent controls. Representative western blots of col type I (A, C) or α-SMA (B, D) are shown. The bottom graphs show RT-PCR for col type I (A, C) or α-SMA (B, D). (E, F) Fibroblasts were seeded into 12-well plates, each containing a glass coverslip and incubated with CCh (E) or TGF-β1 (F) in the presence or absence of aclidinium. Cells were then fixed, permeabilised and immunostained with anti-α-SMA followed by secondary anti-FITC. Fluorescence images are representative of three independent experiments per condition at ×400 and ×1000 magnification. Each graph represents the mean of three experiments for western blots and eight experiments for RNA experiments. One-way repeated measures analysis of variance (ANOVA): p<0.001 (A−D). Post hoc Bonferroni-Holm tests: *p<0.05 compared with solvent controls; #p<0.05 compared with stimulus.
Immunofluorescence of α-SMA in response to carbachol showed a distinct appearance of numerous bundles of actin microfilaments comparable to the response with TGF-β1 (figure 2E,F). Control fibroblasts showed diffuse irregular cytoplasmic α-SMA staining with no filamentous pattern. The percentage of cells expressing α-SMA microfilaments following carbachol or TGF-β1 exposure was 36 (5)% and 60 (10)%, respectively. Aclidinium 10−7 M significantly reduced carbachol- and TGF-β1-induced α-SMA microfilament formation to 5 (2)% and 15 (3)%, respectively.
Carbachol increases myofibroblast markers by a TGF-β1 common mechanism
Carbachol-induced collagen type I and α-SMA protein expression was suppressed when cells were pretreated with aclidinium, the Rho kinase inhibitor Y27632, the cAMP analogue dbcAMP or the ERK1/2 inhibitor PD98059 (figure 3A,B). Western blot analysis showed that carbachol-induced collagen type I and α-SMA expression was almost completely suppressed by an anti-TGF-β1 antibody (4 μg/ml) (figure 3A,B). Furthermore, carbachol increased TGF-β1 cellular protein and mRNA expression (figure 3C–E) and increased a small but significant amount of extracellular TGF-β1 (figure 3F). Pretreatment of cells with aclidinium or with Y27632, dbcAMP or PD98059 effectively suppressed TGF-β1 protein and mRNA expression as well as TGF-β1 extracellular secretion, even below control levels.

Carbachol (CCh)-induced expression of collagen type I (col type I) and α-smooth muscle actin (α-SMA) is partially mediated by transforming growth factor-β1 (TGF-β1). Human lung fibroblasts were stimulated with CCh for 48 h. (A, B) Aclidinium (ACL), Rho kinase inhibitor (Y27632), dibutyryl cyclic AMP analogue (dbcAMP), ERK1/2 inhibitor (PD98059), anti-TGF-β1 antibody (4 μg/ml) or their IgG isotypes were added to the medium 30 min before CCh stimulation. After incubation, total protein was extracted and western blots were performed for col type I (A) and α-SMA (B). Graphs from (A) and (B) represent values of densitometry of col type I (A) and α-SMA (B) protein expression relative to β-actin and normalised to solvent controls. Representative western blots show col type I (A) and α-SMA (B) protein expression. In other experiments, cells were stimulated with CCh in the presence or absence of ACL (C, E, F), Y27632 (D, E, F), PD98059 (D, E, F) or dbcAMP (D, E, F). After incubation, total protein (C, D), RNA (E) or cell culture supernatants were collected and TGF-β1 protein (C, D), mRNA (E) as well as the active form of TGF-β1 present in the culture medium (F) were quantified by western blot, RT-PCR and ELISA, respectively. Each graph represents the mean of three (A−D) or six (E, F) experiments. One-way repeated measures analysis of variance (ANOVA): p<0.001 (A−F). Post hoc Bonferroni-Holm tests: *p<0.05 compared with solvent controls; #p<0.05 compared with stimulus.
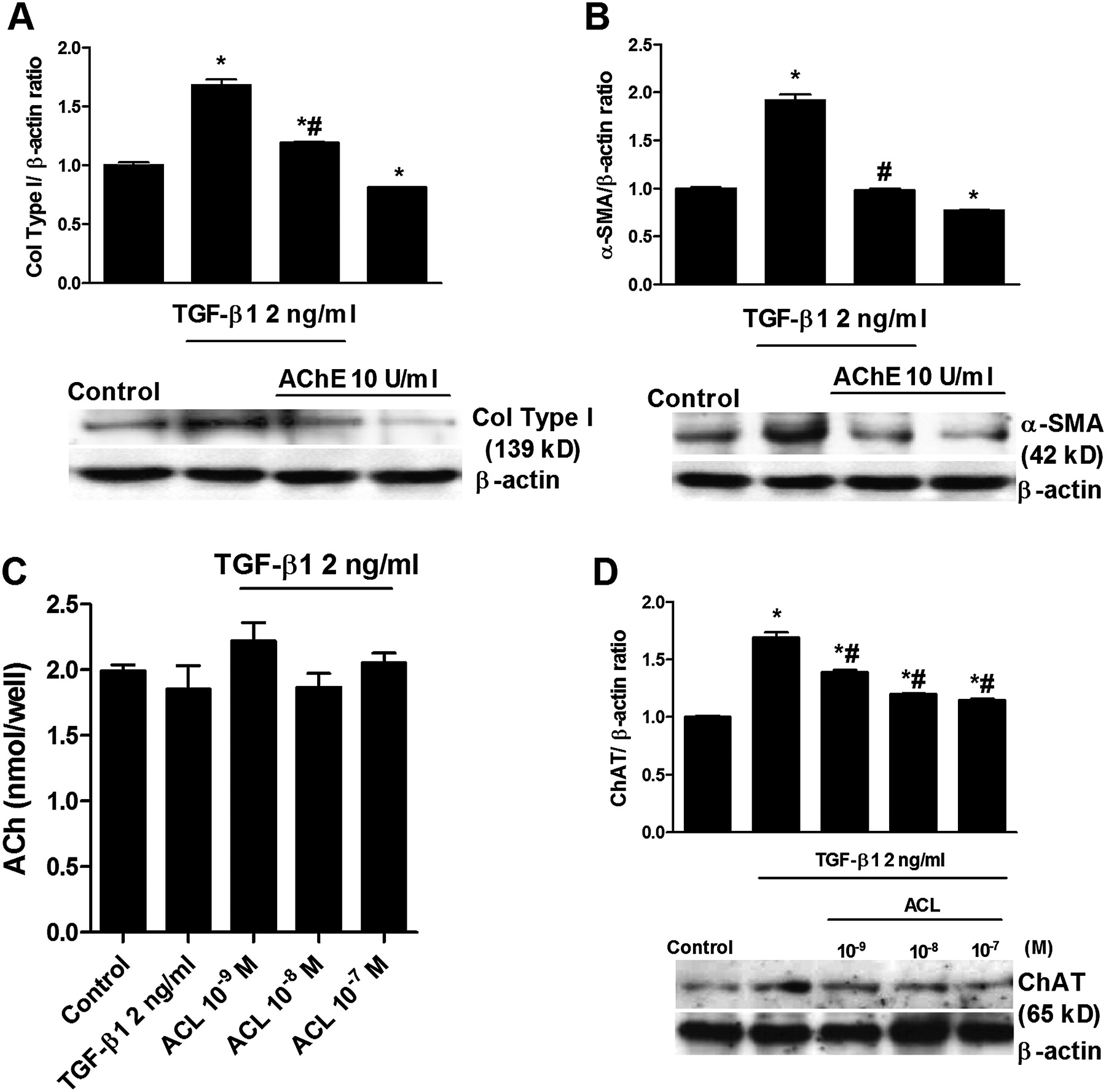
We next explored whether TGF-β1 activates the non-neuronal cholinergic system. The enzyme AChE (10 U/ml) was used to remove any extracellular ACh during the 48-hour period of TGF-β1 stimulation. AChE reduced the increase in collagen type I and α-SMA expression induced by TGF-β1 (figure 4A,B). However, the analysis of ACh supernatant levels revealed no differences between the control group and cells treated with TGF-β1 or aclidinium (figure 4C), probably due to the rapid ACh degradation by extracellular cholinesterases present on fibroblasts.21 22 In other experiments we observed that TGF-β1 is able to upregulate ChAT expression in human lung fibroblasts and that this effect is reversed by aclidinium (figure 4D). All these results suggest that muscarinic receptor activation may share common downstream pathways with TGF-β1. This hypothesis is supported by the inhibition of both carbachol- and TGF-β1-induced increases in ERK1/2 phosphorylation and RhoA-GTP formation by aclidinium (figure 5A,B). Aclidinium also prevented the inhibition of isoprenaline-induced intracellular cAMP levels by carbachol (figure 5C).
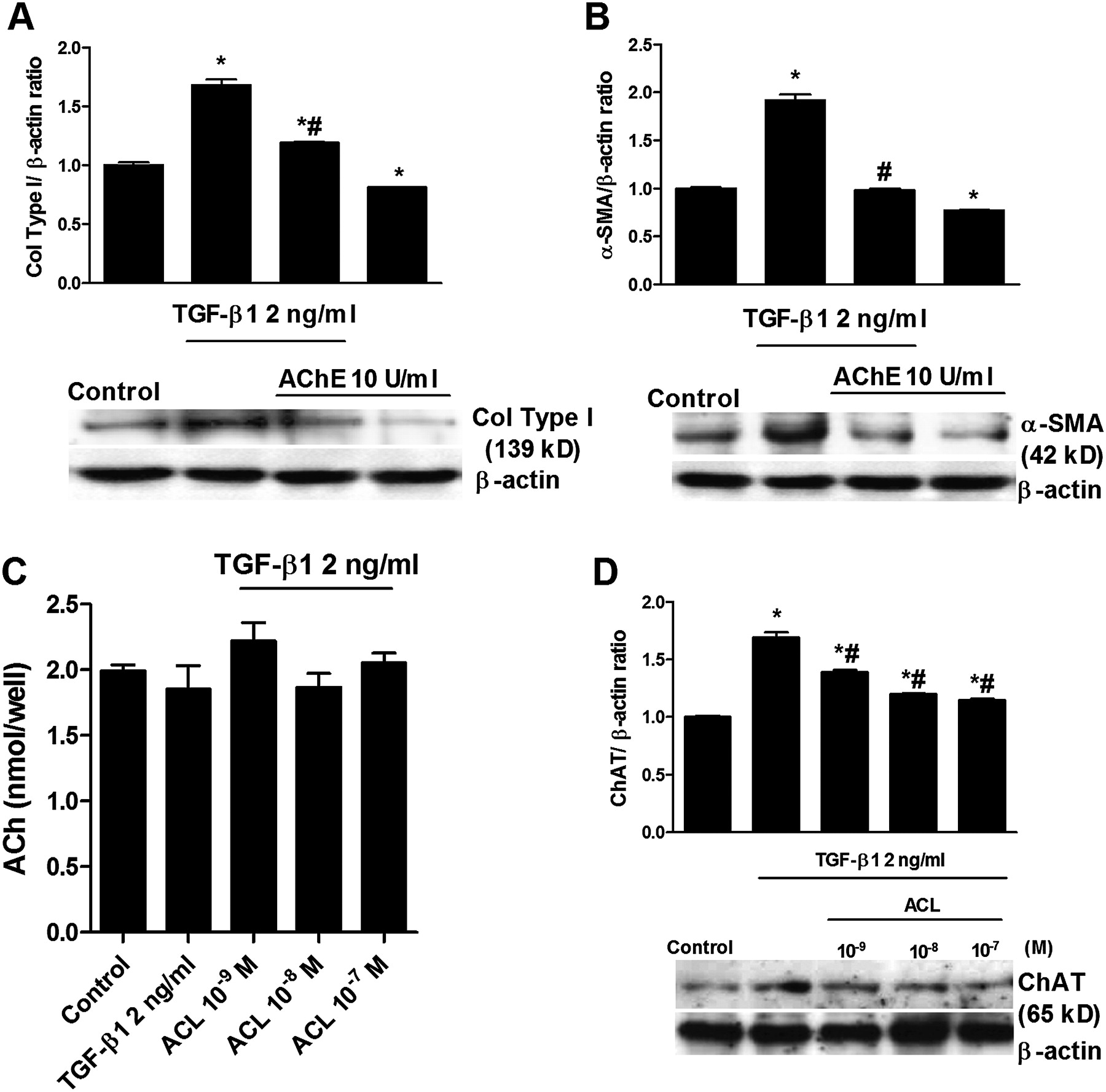
Transforming growth factor-β1 (TGF-β1)-induced expression of collagen type I (col type I) and α-smooth muscle actin (α-SMA) is partially mediated by the non-neuronal acetylcholine system. Human lung fibroblasts were stimulated with TGF-β1 for 48 h. (A, B) Acetyl cholinesterase was added to the medium 30 min before TGF-β1 stimulation. After incubation, total protein was extracted and western blots were performed for col type I and α-SMA. The graphs in (A) and (B) represent values of densitometry of col type I and α-SMA protein expression relative to β-actin and normalised to solvent controls, respectively. Representative western blots show col type I (A) and α-SMA (B) protein expression. In other experiments (C, D), cells were stimulated with TGF-β1 for 48 h in the presence or absence of aclidinium (ACL). After incubation, cell culture supernatants (C) and total protein (D) were collected. Acetylcholine (ACh) supernatant levels were measured by ELISA (C), and choline acetyltransferase (ChAT) expression (D) was quantified by western blot analysis, related to β-actin and normalised to solvent controls. Each graph represents the mean of three (A−D) individual experiments. One-way repeated measures analysis of variance (ANOVA): p<0.001 (A−D). Post hoc Bonferroni-Holm tests: *p<0.05 compared with solvent controls; #p<0.05 compared with stimulus.

Carbachol (CCh)-induced ERK1/2 phosphorylation, RhoA-GTP expression and cAMP inhibition are prevented by aclidinium (ACL). Human lung fibroblasts were stimulated with CCh or transforming growth factor-β1 (TGF-β1) for 48 h (A, B). ACL was added to the medium 30 min before CCh or TGF-β1 stimulation (A, B). After incubation, total protein was extracted and western blots for pERK1/2, total ERK1/2 (A) and RhoA-GTP, RhoA (B) were performed. (C) ACL was incubated for 30 min followed by the addition of CCh for 10 min and isoprenaline (ISO) (10−6 M) for another 10 min. Total cAMP content was determined using biotrack enzyme immunoassay kit and absorbance was read at 450 nm. The Western blots (A, B) are representative of three independent experiments. (C) represents the mean of three individual experiments. One-way repeated measures analysis of variance (ANOVA): p<0.001 (C). Post hoc Bonferroni-Holm tests: *p<0.05 compared with solvent controls; #p<0.05 compared with ISO; ◆p<0.05 related to CCh plus ISO.
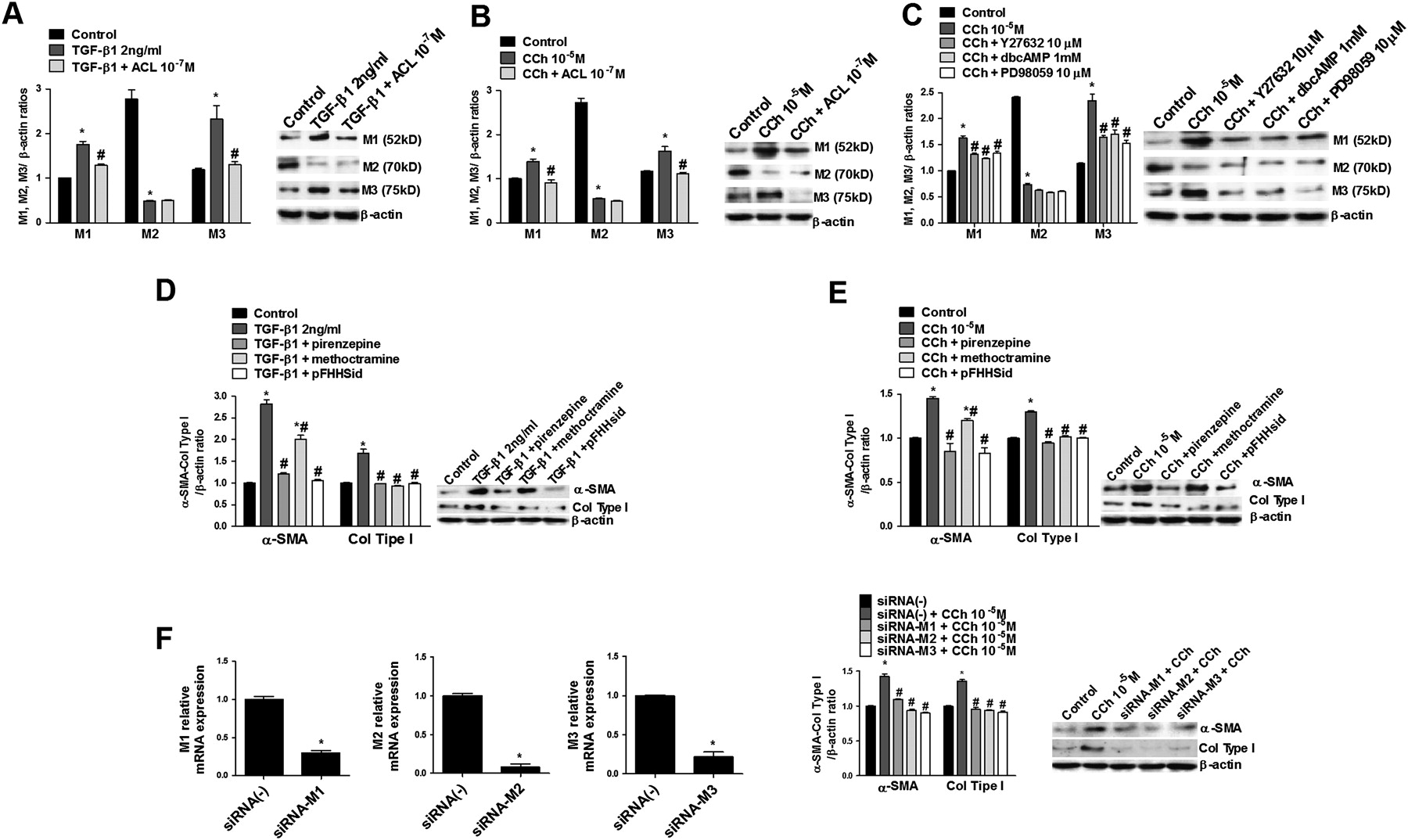
Carbachol and TGF-β1 modify muscarinic receptor expression
Human lung fibroblasts showed high levels of mRNA for M2 transcripts and, to a lesser extent, for M1 and M3 transcripts, respectively (data not shown), while M4 and M5 transcripts were not detected, consistent with previous reports in primary human lung fibroblasts.10 Setting the protein expression level of M1 at 1, the amount of M2 protein expression in human lung fibroblasts under basal conditions was approximately 2.6-fold and 2.4-fold higher than that of M1 and M3, respectively (figure 6A−C). When cells were incubated with TGF-β1or carbachol for 48 h, protein expression of M1 and M3 receptors was upregulated while M2 expression was downregulated (figure 6A,B). Aclidinium pretreatment prevented the upregulation of M1 and M3, but not M2 downregulation induced by carbachol or TGF-β1 (figure 6A,B). In parallel experiments, Y27632, dbcAMP or PD98059 reduced the carbachol-induced M1 and M3 upregulation but did not modify M2 expression (figure 6C).

Carbachol (CCh) and transforming growth factor β1 (TGF-β1) alter muscarinic receptor (M) expression and induce fibroblast to myofibroblast transition via M1, M2 and M3. Human lung fibroblasts were stimulated with TGF-β1 (A) or CCh (B, C) for 48 h. (A, B) aclidinium (ACL), (C) Rho kinase inhibitor (Y27632), (C) dibutyryl cyclic AMP analogue (dbcAMP) or ERK1/2 inhibitor (PD98059) were added to the medium 30 min before stimulation. After incubation, total protein was extracted and western blots were performed for M1, M2 and M3. The graphs (A, B, C) represent values of densitometry of M1, M2 or M3 protein expression relative to β-actin and normalised to solvent controls; representative western blots are also shown for M1, M2 and M3. In other experiments, cells were stimulated with TGF-β1 (D) or CCh (E) in the presence or absence of M1 antagonist pirenzepine, M2 antagonist methoctramine or M3 antagonist p-fluoro-hexahydro siladifenidol (pFHHSid) for 48 h. After incubation, total protein was extracted and western blots were performed for collagen type I (col type I) and α-smooth muscle actin (α-SMA). The graphs (D, E) represent values of densitometry of col type I and α-SMA protein expression relative to β-actin and normalised to solvent controls; representative western blots are shown for col type I and α-SMA. (F) Left black graphs show M1, M2 and M3 mRNA expression after 48 h of treatment with siRNA-M1, M2 and M3. Right graph (F) shows cells selectively transfected with siRNA-M1, M2 and M3 that were stimulated with CCh for 48 h. After incubation, total protein was extracted and col type I and α-SMA protein expression were expressed relative to β-actin and normalised to solvent controls. Representative western blots are shown for col type I and α-SMA. Each graph represents the mean of three (A−F) individual experiments. One-way repeated measures analysis of variance (ANOVA): p<0.001 (A−F). Post hoc Bonferroni-Holm tests: *p<0.05 compared with solvent controls; #p<0.05 compared with stimulus.
To better understand which muscarinic receptor is involved in myofibroblast transition, fibroblasts were preincubated with the M1 antagonist pirenzepine, M2 antagonist methoctramine or M3 antagonist pFHHSid. Both collagen type I and α-SMA expression increased by carbachol or TGF-β1 were prevented by M1, M2 and M3 antagonists, although the M2 antagonist methoctramine was less effective at inhibiting α-SMA expression (figure 6D,E). In other experiments in which M1, M2 and M3 receptors were selectively silenced by transfecting cells with siRNA-M1, siRNA-M2 and siRNA-M3, carbachol-induced collagen type I and α-SMA were totally suppressed (figure 6F).
Aclidinium inhibits common phenotypic alterations of myofibroblasts
Subconfluent cells were exposed to TGF-β1 or carbachol for 48 h in the presence or absence of aclidinium (10−9 to 10−7 M). Both TGF-β1 and carbachol increased cell proliferation by approximately 2.25- and 1.75-fold, respectively (figure 7A−C). Aclidinium dose-dependently inhibited the TGF-β1 and carbachol-induced cell proliferation with statistical significance at 10−8 M and 10−7 M (figure 7A,B). Furthermore, Y27632, dbcAMP and PD98059 preincubation effectively reduced cell proliferation induced by carbachol (figure 7C). In other experiments, human bronchial fibroblast closure studies were performed to analyse cell migration. Aclidinium (figure 7D,E) and Y27632, dbcAMP and PD98059 (figure 7F) dose-dependently inhibited cell migration and wound closure.

Aclidinium inhibits fibroblast proliferation and migration. Semiconfluent fibroblasts were grown in 96-well plates in the presence or absence of transforming growth factor β1 (TGF-β1) (A) or carbachol (CCh) (B) for 48 h. (A, B) Aclidinium (ACL), (C) Rho kinase inhibitor (Y27632), dibutyryl cyclic AMP analogue (dbcAMP) or ERK1/2 inhibitor (PD98059) were added to the medium 30 min before stimulation. After incubation, cell proliferation was assessed by colorimetric BrDU assay. In panels (D−F), fibroblasts were stimulated for 48 h with CCh or vehicle (control) in the presence or absence of ACL (D, E), Y27632 (F), dbcAMP (F) or PD98059 (F). After stimulation, a circular wound edge was created in the centre of the well by removing a p-200 pipette tip. The cells were washed and re-exposed to the same conditions and wound closure was monitored for 24 h and 48 h. (D) Circular wound closure image at ×50 final augmentation (scale bar 60 μm). Each graph represents the mean of three individual experiments. One-way repeated measures analysis of variance (ANOVA): p<0.001 (A−F). Post hoc Bonferroni-Holm tests: *p<0.05 compared with solvent controls; #p<0.05 compared with stimulus.
Discussion
This study describes for the first time the mechanistic and functional involvement of a non-neuronal cholinergic system in the lung fibroblast to myofibroblast transition and provides evidence for the use of anticholinergic agents in lung diseases in which lung remodelling occurs. The relevance of these findings was highlighted by the finding that aclidinium effectively attenuated carbachol- and TGF-β1-induced myofibroblast transition in bronchial fibroblasts from smokers and patients with COPD. These observations suggest that aclidinium may have anti-remodelling properties in addition to its sustained bronchodilation and positive safety profile for anticholinergic adverse events observed in clinical trials.23
Myofibroblasts are characterised by secretion of extracellular matrix components such as collagen type I (a characteristic shared with fibroblasts) and by formation of contractile apparatus such as α-SMA microfilaments (a characteristic shared with airway smooth muscle cells). Furthermore, myofibroblasts show a high proliferative and migratory activity under inflammatory conditions.3 In this respect, recent human in vivo evidence has shown that muscarinic agonist (methacholine) inhalation promotes myofibroblast expansion and airway smooth muscle dedifferentiation into myofibroblasts that migrate to the submucosal level.24 These data, together with the capacity of ACh to promote fibroblast proliferation and collagen secretion, suggest that the cholinergic system may be involved in the process of fibroblast to myofibroblast transition and contribute to the increased pool of myofibroblasts that has been observed in the lungs of patients with asthma and COPD.
Until now, available data of muscarinic agonist effects on lung fibroblasts have been restricted to fibroblast proliferation and collagen synthesis.10 11 25 In this study we show for the first time that collagen type I and α-SMA microfilaments are upregulated following carbachol exposure. Furthermore, we demonstrate a link between the cholinergic and TGF-β1 systems. Previous reports showed that cholinergic receptor stimulation may increase the expression and release of TGF-β in different cell types.26–28 Recently it was observed that lung fibroblasts stimulated with interleukin 1β (IL-1β), tumour necrosis factor α (TNFα) or cigarette smoke extract increase ChAT expression, suggesting the presence of a non-neuronal ACh system in lung fibroblasts.9 In our study, carbachol increased TGF-β1 expression and secretion, which may account for the induction of collagen type I and α-SMA which was inhibited by an anti-TGF-β1 antibody. Furthermore, aclidinium was able to significantly reduce the TGF-β1 expression and secretion induced by carbachol, which indicates that muscarinic receptors are also involved in this process. Similar to what occurs under inflammatory conditions, TGF-β1 was able to increase ChAT expression and, more interestingly, the addition of AChE suppressed the TGF-β1-induced increase in collagen type I and α-SMA. These results suggest that TGF-β1 activates a non-neuronal cholinergic system.
Since carbachol and TGF-β1 may alter cell markers to induce fibroblast to myofibroblast transition, changes in the expression of M receptors may also be expected. The pattern of M receptor expression on lung fibroblasts has recently been studied in detail,29 but data on the expression of myofibroblast M receptors are scarce. It has been reported that human lung fibroblasts express high amounts of M2 mRNA transcripts and protein followed by M1 and, to a lesser extent, M3,9 29 which is in agreement with our results (figure 6). Interestingly, both carbachol and TGF-β1 increased M1 and M3 protein expression and reduced M2 expression after 48 h of exposure. Similar findings were also shown in lung fibroblasts following exposure to IL-1β, TNFα or cigarette smoke extract.9 Furthermore, lung fibroblasts from patients with COPD showed an upregulation of M1 and M3 and downregulation of M2 with respect to control subjects.9 Previous reports also showed that TGF-β1 downregulates M2 expression in human embryonic lung fibroblasts30 and may increase M3 expression in smooth muscle cells.31 Since the myofibroblast phenotype possesses morphological and biochemical features intermediate between fibroblasts and smooth muscle cells, the M1 and M3 upregulation found after myofibroblast transition could be related to their high contractile cellular capacity, as shown previously.32 These results are consistent with the in vivo conditions where M3 and, to a lesser extent, M1 are coupled to Gq protein and therefore contribute to bronchial and tracheal smooth muscle contraction.8 We also found that the antimuscarinic aclidinium attenuates M1 and M3 upregulation induced by carbachol and TGF-β1, which in turn correlates with the inhibitory effect of aclidinium on fibroblast to myofibroblast transition.
It remains unclear whether there is a specific muscarinic receptor involved in fibroblast to myofibroblast transition. Until now, M2 was considered to be the main receptor subtype responsible for fibroblast proliferation and collagen synthesis. These data were based on the high M2 expression and by the action of the Gi/o protein inhibitor pertussis toxin and M-subtype antagonists with a limited selective action.10 11 Results from lung fibroblasts recently showed that all muscarinic receptors are involved in fibroblast proliferation.9 In the present study we further evaluate this thesis by using pharmacological intervention (M1, M2 and M3 antagonists) as well as genetic techniques that allow specific silencing of each receptor (siRNAs of M1, M2 and M3). We demonstrated that collagen type I and α-SMA upregulation—and therefore myofibroblast transition—is mediated by these three types of muscarinic receptors.
There is also evidence of ERK1/2 involvement on the muscarinic agonist-induced fibroblast proliferation and collagen synthesis.9 10 25 In the present study we found that carbachol and TGF-β1 induce ERK1/2 phosphorylation, and that this effect was effectively blocked by aclidinium. Furthermore, PD98059, the specific inhibitor of ERK1/2, attenuates carbachol-induced collagen type I and α-SMA upregulation, cell proliferation and migration, as well as TGF-β1 expression and secretion. Similar results were observed for RhoA activation and cAMP downregulation induced by carbachol. It is known that muscarinic receptors and TGF-β1 activate ERK1/2 and RhoA-GTP.8 28 RhoA activation has been implicated in fibroblast proliferation and migration,33 and increased intracellular cAMP levels act as a potent inhibitor of the fibroblast to myofibroblast transition.34 In this study, aclidinium was shown to inhibit RhoA-GTP formation and prevent carbachol-induced cAMP downregulation. Interestingly, the inhibitors of Rho kinase (Y27632 and dbcAMP) were able to inhibit fibroblast to myofibroblast transition as well as cell proliferation and migration. These data support ERK1/2, RhoA-GTP and cAMP as intracellular messengers that contribute to carbachol-induced fibroblast to myofibroblast transition (figure 8). Furthermore, the muscarinic antagonist aclidinium inhibits the activation of ERK1/2 and RhoA-GTP by carbachol and TGF-β1, and prevents the decrease of cAMP levels, which in turn inhibits the myofibroblast transition (figure 8).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Model to explain the transforming growth factor β1 (TGF-β1)-induced non-neuronal cholinergic system on bronchial fibroblast to myofibroblast transition as well as the inhibitory effect of aclidinium (ACL). The muscarinic receptor agonist carbachol (CCh) increases the myofibroblast markers collagen type I (Col type I) and α smooth muscle actin (α-SMA) through the phosphorylation of extracellular signal-regulated protein kinase (ERK)-1/2 and activation of Rho kinase. Depletion of intracellular cyclic AMP (cAMP) induced by CCh also contributes to Col type I and α-SMA upregulation. CCh activates the expression and secretion of TGF-β1 which activates the choline acetyltransferase (ChAT) expression and activity in an autocrine fashion. ChAT increases the synthesis and secretion of acetylcholine (ACh) which activates the muscarinic receptors and promotes fibroblast to myofibroblast transition in an autocrine fashion. The antimuscarinic aclidinium (ACL) inhibits the effects of CCh and ACh on fibroblast to myofibroblast transition. Thus, ACL inhibits ERK1/2 phosphorylation, Rho kinase activation, cAMP downregulation and Col type I and α-SMA expression. Furthermore, ACL inhibits TGF-β1 expression and secretion as well as the non-neuronal cholinergic activation of fibroblast to myofibroblast transition.
In addition to the pathways explored in this work, numerous previous studies have shown that TGF-β1 induces myofibroblast differentiation including collagen and α-SMA expression via Smad-mediated pathways. It has recently been shown that the anticholinergic tiotropium may suppress TGF-β1-induced Smad2/4 activation in lung fibroblasts suggesting a crosstalk between TGF-β1 and muscarinic signalling.35
In summary, we have shown that activation of muscarinic receptors induces fibroblast to myofibroblast transition through an increase in ERK1/2 phosphorylation, RhoA-GTP formation and downregulation of cAMP levels and provide evidence that cholinergic and TGF-β1 pathways interact to induce myofibroblast transition. The muscarinic antagonist aclidinium was a potent inhibitor of the fibroblast to myofibroblast transition following carbachol or TGF-β1 exposure. We therefore conclude that, in addition to its bronchodilatory activity, aclidinium may play a role in regulating fibrotic remodelling in chronic inflammatory diseases such as asthma and COPD.
Acknowledgments
We are grateful for the valuable help of Professors M Guijarro and M Juan of the Medicine Department of the Valencia University General Hospital for access to, and isolation of, human fibroblasts. We thank Clare Smith from Complete Medical Communications who provided editing assistance funded by Almirall SA, Barcelona, Spain.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
JM and AS contributed equally to this work.
Funding This work was supported by Almirall SA, Barcelona, Spain and in part by grants SAF2008-03113 (JC), SAF2009-08913 (EJM), CIBERES (CB06/06/0027) and research grants from Regional Government (Prometeo/2008/045, ‘Generalitat Valenciana’; Emerging Groups GE-029/10, ‘Generalitat Valenciana’(to JM)). Support from the CENIT programme (Spanish government) and Almirall (Barcelona, Spain) was obtained. Almirall SA was not involved in the study design, collection, analysis or interpretation of data, writing of the report or the decision to submit the paper for publication. TP received a research grant from Conselleria de Educación, Generalitat Valenciana ACIF/2010/114.
Competing interests EJM and JC received a research grant from Almirall. AG and MM are employees of Almirall.
Patient consent Obtained.
Ethics approval This study has been approved by the ethics committee of the General Hospital of Valencia, Spain.
Provenance and peer review Not commissioned; externally peer reviewed