Article Text

Abstract
Background Idiopathic pulmonary fibrosis (IPF) is characterised by the aberrant epithelial to mesenchymal transition (EMT) and myofibroblast accumulation. Sphingosine-1-phosphate (S1P) and sphingosine kinase 1 (SPHK1) have been implicated in lung myofibroblast transition, but their role in EMT and their expression in patients with IPF is unknown.
Methods and results S1P levels were measured in serum (n=27) and bronchoalveolar lavage (BAL; n=15) from patients with IPF and controls (n=30 for serum and n=15 for BAL studies). SPHK1 expression was measured in lung tissue from patients with IPF (n=12) and controls (n=15). Alveolar type II transformation into mesenchymal cells was studied in response to S1P (10−9–10−5 M). The median (IQR) of S1P serum levels was increased in patients with IPF (1.4 (0.4) μM) versus controls (1 (0.26) μM; p<0.0001). BAL S1P levels were increased in patients with IPF (1.12 (0.53) μM) versus controls (0.2 (0.5); p<0.0001) and correlated with diffusion capacity of the lung for carbon monoxide, forced expiratory volume in 1 s and forced vital capacity (Spearman's r=−0.87, −0.72 and −0.68, respectively) in patients with IPF. SPHK1 was upregulated in lung tissue from patients with IPF and correlated with α-smooth muscle actin, vimentin and collagen type I (Spearman's r=0.82, 0.85 and 0.72, respectively). S1P induced EMT in alveolar type II cells by interacting with S1P2 and S1P3, as well as by the activation of p-Smad3, RhoA-GTP, oxidative stress and transforming growth factor-β1 (TGF-β1) release. Furthermore, TGF-β1-induced EMT was partially conducted by the S1P/SPHK1 activation, suggesting crosstalk between TGF-β1 and the S1P/SPHK1 axis.
Conclusions S1P is elevated in patients with IPF, correlates with the lung function and mediates EMT.
- Sphingosine-1-phosphate
- sphingosine kinase 1
- epithelial to mesenchymal transition
- idiopathic pulmonary fibrosis
- airway epithelium
- interstitial fibrosis
- oxidative stress
Statistics from Altmetric.com
- Sphingosine-1-phosphate
- sphingosine kinase 1
- epithelial to mesenchymal transition
- idiopathic pulmonary fibrosis
- airway epithelium
- interstitial fibrosis
- oxidative stress
Key messages
What is the key question?
Is the bioactive lipid, sphingosine-1-phosphate (S1P), involved in the idiopathic pulmonary fibrosis (IPF) process?
What is the bottom line?
S1P and sphingosine kinase 1 (SPHK1) are upregulated in serum, bronchoalveolar lavage and lung tissue of patients with IPF, correlate with lung function and mediate epithelial to mesenchymal transition.
Why read on?
This is the first report that relates S1P and SPJK1 with the IPF process and with the epithelial to mesenchymal transition as an underlying cellular process. The data presented in this study could be of potential value in future antifibrotic therapeutic interventions focused on the S1P system.
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common idiopathic interstitial pulmonary disease. Owing to its poor prognosis and aggressive nature, IPF poses major challenges to clinicians.1 Currently, no effective treatments exist to stop ongoing fibrosis in IPF. The accumulation and persistence of myofibroblasts is believed to contribute to the development of fibrosis.2 α-Smooth muscle actin (α-SMA) and vimentin expression, increased proliferative capacity, and increased generation and secretion of extracellular matrix (ECM) proteins such as collagen and fibronectin are key hallmarks of myofibroblast differentiation in fibrotic disorders.2 It has been established that myofibroblast foci may develop as a consequence of fibroblast/myofibroblast transition, alveolar epithelial to mesenchymal transition (EMT), or recruitment of circulating fibroblastic stem cells (fibrocytes).2 The formation and progression of myofibroblasts occurs mainly through combinatorial signals involving transforming growth factor β1 (TGF-β1) and integrin signalling.3 However, it was recently shown that other growth factors such as platelet-derived growth factor and molecules such as the sphingolipid sphingosine-1-phosphate (S1P) may activate lung fibroblasts/myofibroblasts.4 The bioactive phospholipid S1P has been presented as a lung pro-inflammatory/pro-remodelling agent because S1P levels are increased in bronchoalveolar lavage (BAL) fluid from patients with asthma after antigen challenge and bleomycin-induced pulmonary fibrosis in animal models.5 6 Most of the S1P effects are mediated through members of the G-protein-coupled S1P receptor family, which includes ubiquitously expressed subtypes S1P1, S1P2 and S1P3.7 Interestingly, the main pro-fibrotic factor TGF-β1 activates and upregulates sphingosine kinase 1 (SPHK1),4 the enzyme that catalyses phosphorylation of sphingosine to produce S1P. Thus, SPHK1 establishes a link between TGF-β1 and S1P to promote common pro-fibrotic actions by means of S1P receptor transactivation.4 Despite in vitro evidence of S1P and SPHK1 in lung fibroblast activation, there is no evidence of either the effect of S1P on the EMT process or the presence and distribution of S1P and SPHK1 in patients with IPF. Therefore, we hypothesised that S1P and SPHK1 could be over-expressed in patients with IPF and that S1P could mediate EMT in human alveolar epithelial cells, which in turn may contribute to lung fibrosis formation and progression.
The aim of this study was to investigate S1P and SPHK1 expression in serum, BAL and lung tissue from controls and patients with IPF and to analyse the effect of S1P on alveolar EMT as a new myofibroblast inducer.
Materials and methods
Patients with IPF
A total of 27 patients with IPF were included in the study. IPF was diagnosed according to the American Thoracic Society/European Respiratory Society (ATS/ERS) consensus criteria.8 Fibrotic lung samples were obtained by open lung biopsy. Serum samples were obtained from peripheral blood, and alveolar macrophages were obtained and isolated from BAL as described below.
Controls
Lung tissue samples were obtained from patients undergoing thoracic surgery for removal of a primary lung tumour. Normal lung was obtained from a non-involved segment, remote from the solitary lesion. Serum samples were obtained from healthy subjects without any medical disease. Control BAL and control alveolar macrophages were obtained from patients who were undergoing bronchoscopy as a diagnostic approach to haemoptysis, which in all cases showed no macroscopic lesions; further, no radiological anomalies were shown on CT. To investigate whether airways were colonised with bacteria, bronchial secretions were obtained using a protected specimen brush. These were found to be culture negative in all cases. The protocol was approved by the local research and independent ethics committee of the University General Hospital of Valencia. Informed written consent was obtained from each participant.
Cell culture and stimulation
Alveolar macrophages were isolated from BAL fluid by means of adherence properties on a culture cell plate. Human alveolar type II cells (ATII) were isolated from human lung tissue as described previously, with modifications (see online supplement).9 The A549 cells were purchased from American Type Culture Collection (Rockville, Maryland, USA). Stimulation conditions and inhibitors are defined in the online supplement.
Proliferation assay
Cell proliferation was measured by colorimetric immunoassay based on BrdU incorporation during DNA synthesis using a cell proliferation ELISA BrdU kit (Roche, Mannheim, Germany) according to the manufacturer's protocol, as previously outlined.10
Detection of RhoA-GTP, S1P, total soluble collagen, and supernatant TGF-β1
Analysis of intracellular RhoA-GTP, S1P, soluble collagen and TGF-β1 was performed using a RhoA-GTP activity assay kit (G-LISA; Cytoskeleton, Denver, Colorado, USA), S1P competitive ELISA kit (Echelon Biosciences Inc., Salt Lake City, Utah, USA), Sircol assay kit (Biocolor, Belfast, Ireland) and Quantikine Human TGF-β1 Immunoassay (catalogue no. 891124; R&D Systems). See online supplement for further details.
Real-time PCR
Real-time PCR was performed as previously described.11 Relative quantification of different transcripts was determined by the 2−ΔΔCt method, using glyceraldehydes-3-phosphate dehydrogenase (GAPDH) as an endogenous control and with normalisation to the control group. See online supplement for further details.
Protein array
Protein expression in lung tissue samples from controls and patients with IPF was analysed with Zeptosens (Division of Bayer (Schweiz), Switzerland) protein array technology as previously outlined.12 See online supplement for further details.
Western blot analysis
Protein expression in A549 cells was analysed by western blot as previously described.11 Primary antibodies against SPHK1, TGF-β1, α-SMA, vimentin, E-cadherin and p-Smad were used. See online supplement for further details.
Immunohistochemistry
SPHK1 immunohistochemical analysis of human pulmonary tissue was performed using rabbit anti-human SPHK1 antibody (1:100; Sigma, Madrid, Spain). Details are described in the online supplement.
DCFDA fluorescence measurement of reactive oxygen species
Intracellular reactive oxygen species (ROS) levels (H2O2 and superoxide anion) were measured in A549 cells by means of dichlorofluorescin diacetate (DCFDA) dye as previously outlined.11 See online supplement for further details.
Statistics
Statistical analysis of the results was carried out by parametric or non-parametric analysis as appropriate. Significance was accepted when p<0.05. Details are described in the online supplement.
Results
S1P levels and SPHK1 expression are increased in patients with IPF
Controls and patients with IPF were prospectively recruited from the Respiratory Unit, University General Hospital Consortium, Valencia, Spain between 2008 and 2010 at the initial diagnostic work-up. Clinical data of patients are shown in table 1.
Clinical features of patients
Serum and BAL levels of S1P were significantly higher in patients with IPF than in controls (p<0.0001) (figure 1A,B). The SPHK1 mRNA transcript level was increased in alveolar macrophages of patients with IPF compared with controls (p=0.004) (figure 1C). No correlation was found between the serum S1P levels in patients with IPF and the clinical features or radiological findings. In contrast, BAL S1P levels in patients with IPF were inversely correlated with diffusion capacity of the lung for carbon monoxide (DLco), forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC) (Spearman's r=−0.87 (p=0.0072), −0.72 (p=0.036) and −0.68 (p=0.044), respectively), but not with the extension of radiological findings. S1P levels were not correlated with leucocyte or lymphocyte numbers in BAL fluid.

Sphingosine-1-phosphate (S1P) and sphingosine kinase 1 (SPHK1) are increased in patients with idiopathic pulmonary fibrosis (IPF) compared with controls. S1P concentration was measured by ELISA in (A) serum from 30 healthy controls and 27 patients with IPF, and (B) bronchoalveolar lavage (BAL) fluid from 15 non-pathological controls and 15 patients with IPF. (C) In other experiments, alveolar macrophages were isolated from BAL fluid to quantify SPHK1 mRNA transcripts in 15 non-pathological controls and 15 patients with IPF. The results are expressed as median (IQR) of triplicate determinations for each sample. p Values for comparison by Mann–Whitney test are shown.
Compared with lung tissue from controls, lung tissue from patients with IPF showed higher protein expression levels of fibrotic markers such as α-SMA (p=0.042), vimentin (p=0.012) and collagen (col) type I (p=0.048) and also for SPHK1 (p=0.045; figure 2A). In this regard, SPHK1 protein expression was directly correlated with α-SMA, vimentin and col type I expression in patients with IPF (Spearman's r=0.82 (p=0.041), 0.85 (p=0.013) and 0.72 (p=0.033), respectively). Immunohistochemistry showed that bronchial and alveolar epithelial cells expressed SPHK1 in the normal lung (figure 2B). In IPF lung samples, hyperplastic alveolar cells (figure 2B, red arrows) and fibroblasts (figure 2B, black arrows) were strongly labelled with anti-SPHK1 antibody.

Sphingosine kinase 1 (SPHK1) is overexpressed in idiopathic pulmonary fibrosis (IPF) human lung tissue. (A) Non-pathological lung tissue and IPF lung tissue were homogenised to extract proteins. Protein expression of α-smooth muscle actin (α-SMA), vimentin, collagen (col) type I, SPHK1 and β-actin (as internal control) was analysed by protein array Zeptosens technology with specific antibodies. A total of 15 control and 12 IPF lung samples were included in the experiments. The upper graph shows protein expression corrected using the internal control (β-actin) and normalised to control protein expression. On the right is a representative picture of the protein array. p Values for comparison by Mann–Whitney test are shown. (B) Detection of immunoreactive SPHK1 in the lung. Immunohistochemistry shows that bronchial and alveolar epithelial cells express SPHK1 in the normal lung. In IPF lung samples, hyperplastic alveolar cells and fibroblasts are strongly labelled (see arrows). The control antibody always gave a negative signal.
S1P induces alveolar EMT
The ATII epithelial cell line A549 cultured in the absence of S1P maintained classic cobblestone epithelial morphology as assessed by phase contrast light microscopy (figure 3A). A concentration of 10−7 M S1P (72 h) began to induce morphological changes in A549 cells, characterised by a more fibroblast-like morphology with reduced cell–cell contact (figure 3A). In addition, mRNA transcripts of the epithelial cell markers ZO-1 and E-cadherin were significantly downregulated, and mRNA transcripts of the mesenchymal phenotype markers α-SMA, vimentin and col type I were upregulated in a dose-dependent manner after S1P exposure, which confirmed EMT (figure 3B). Protein analysis further confirmed these results for α-SMA, vimentin and E-cadherin (figure 3C) as well as for total soluble collagen (figure 3D). Primary human ATII cells stimulated with 10−6 M S1P for 72 h showed similar changes (figure 3E). The observed changes in cell phenotype were accompanied by the arrest of cell growth after 72 h of S1P stimulation (figure 3F).

Sphingosine-1-phosphate (S1P) induces alveolar epithelial to mesenchymal transition (EMT). Human type II epithelial alveolar cells (ATII) and A549 cells were stimulated in the presence or absence of S1P for 72 h. (A) Morphological changes in A549 cells began at a S1P concentration of 10−7 M, which induced a more fibroblast-like morphology with reduced cell–cell contact as assessed by phase contrast light microscopy. Scale bar: 10 μm. (B) S1P dose-dependently upregulated mRNA transcripts of the mesenchymal markers α-smooth muscle actin (α-SMA), vimentin and collagen (col) type I and downregulated mRNA transcripts of the epithelial markers E-cadherin and ZO-1. (C) S1P dose-dependently increased the protein levels of the mesenchymal markers α-SMA, vimentin and (D) soluble collagen, and downregulated the protein level of the (C) epithelial marker E-cadherin. (E) ATII cells were stimulated with 1 μM S1P for 72 h, and the α-SMA, vimentin, col type I, E-cadherin and ZO-1 mRNA transcripts were measured. (F) Sub-confluent A549 cells were cultured in 96-well plates and stimulated with S1P at different concentrations and for different periods of time. Results are expressed as means (SE) of n=4 experiments per condition. Post hoc Bonferroni tests: exact p values down to 0.05 are indicated in the graph; *p<0.05; **p<0.01; ***p<0.001; related to solvent controls.
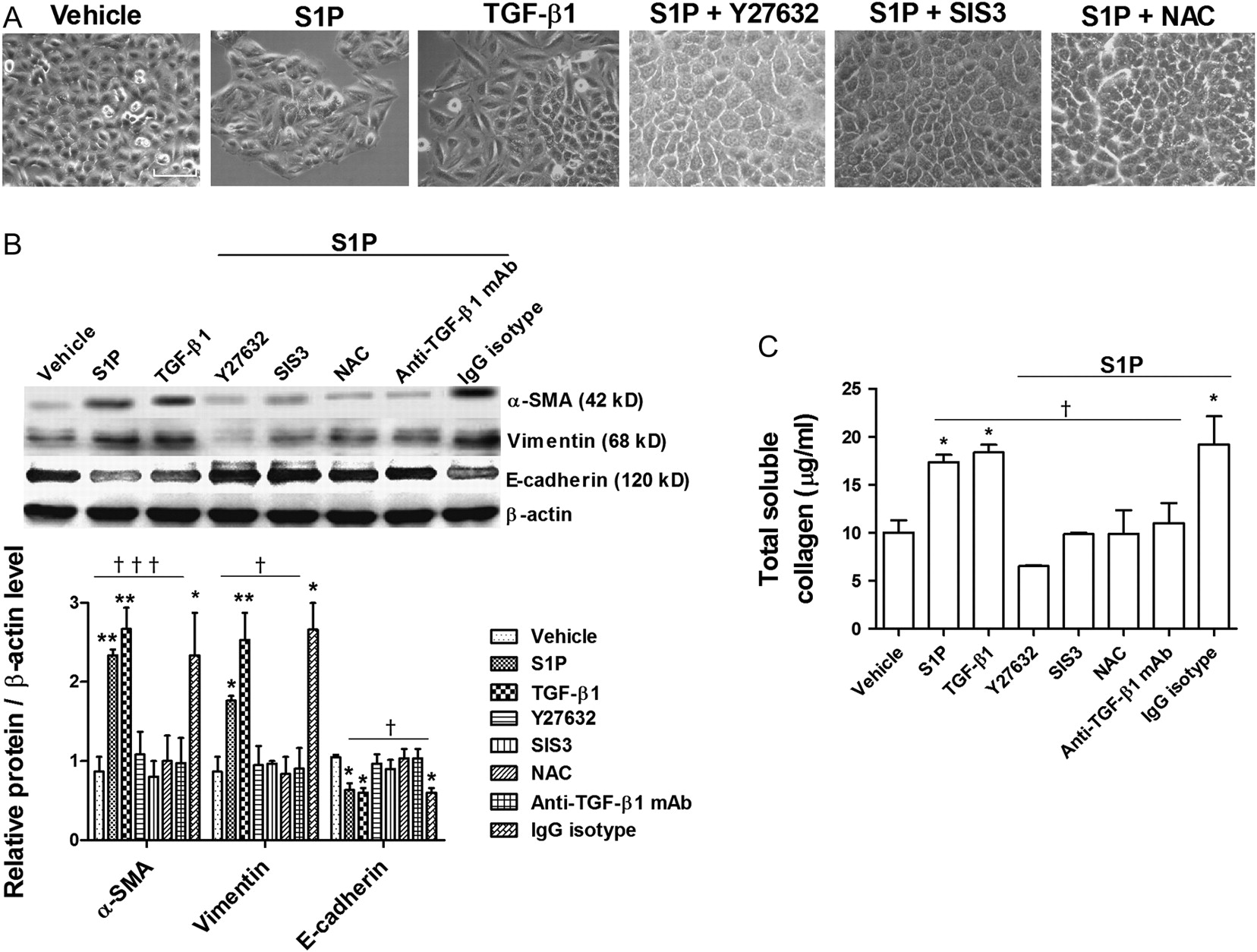
S1P-induced EMT is partially mediated by RhoA-GTP, Smad3, intracellular ROS and autocrine TGF-β1 action
The Rho kinase inhibitor Y27632, Smad3 inhibitor SIS3 and antioxidant N-acetyl-L-cysteine (NAC) suppressed the S1P-induced phenotypic changes in A549 cells (figure 4A) and partially suppressed the increase in α-SMA, vimentin and collagen as well as the decrease in E-cadherin protein expression (figure 4B,C). It was recently suggested that S1P shares common intracellular pathways with TGF-β1 and that S1P may trans-activate TGF-β receptor.13 In the present work, TGF-β1 promoted EMT in a manner similar to that of S1P (figure 4A–C). Furthermore, anti-TGF-β1 antibody inhibited S1P-induced EMT, suggesting an autocrine role for TGF-β1 (figure 4B,C). Specific antagonists for S1P1, S1P2 and S1P3 were used to elucidate the S1P receptor subtypes required for the S1P-induced EMT process. The S1P2 inhibitor JTE013 and the S1P3 inhibitor CAY10444 attenuated S1P-induced α-SMA, vimentin and collagen upregulation and E-cadherin downregulation, whereas the S1P1 inhibitor W146 had no effect (figure 5A,B).

Sphingosine-1-phosphate (S1P)-induced epithelial to mesenchymal transition (EMT) was mediated in part by RhoA-GTP, Smad3, intracellular reactive oxygen species (ROS) and extracellular transforming growth factor-β1 (TGF-β1). A549 cells were incubated with the Rho-kinase inhibitor Y27632 (10 μM), Smad3 inhibitor SIS3 (10 μM), antioxidant N-acetyl-L-cysteine (NAC; 1 mM) or anti-TGF-β1 antibody (4 μg/ml) and its negative control (non-immune IgG) for 30 min before S1P (1 μM) stimulation for 72 h. TGF-β1 (5 ng/ml; 72 h) was used as a positive control for EMT. (A) Representative images were photographed under a phase contrast light microscope. Scale bar: 20 μm. (B) Representative images of three western blots with respective densitometry analyses are shown for the mesenchymal markers α-SMA and vimentin and the epithelial marker E-cadherin. Results were corrected to β-actin as an internal control and normalised to the non-treated vehicle group. (C) Total soluble collagen in cell cultures was measured by the Sircol assay. Results are the means (SE) of three independent experiments. Post hoc Bonferroni tests: exact p values down to 0.05 are indicated in the graph (related to the indicated values or to solvent controls if placed on top of a bar); *p<0.05; **p<0.01 related to solvent controls; †p<0.05; †††p<0.001 related to S1P-stimulated cell values as indicated.

Sphingosine-1-phosphate (S1P)-induced epithelial to mesenchymal transition (EMT) was mediated via S1P2 and S1P3. A549 cells were incubated with the S1P1 inhibitor W146 (1 μM), S1P2 inhibitor JTE013 (1 μM) or S1P3 inhibitor CAY10444 (10 μM) for 30 min before S1P (1 μM) stimulation for 72 h. (A) Representative images of three western blots with respective densitometry analyses are shown for the mesenchymal markers α-smooth muscle actin (α-SMA) and vimentin and the epithelial marker E-cadherin. Results were corrected to β-actin as an internal control and normalised to the non-treated vehicle group. (B) Total soluble collagen in cell cultures was measured by the Sircol assay. Results are the means (SE) of three independent experiments. Post hoc Bonferroni tests: exact p values down to 0.05 are indicated in the graph (related to the indicated values or to solvent controls if placed on top of a bar); *p<0.05; **p<0.01 related to solvent controls; †p<0.05; ††p<0.01 related to S1P-stimulated cell values as indicated.
S1P upregulates Smad3 phosphorylation, RhoA-GTP activation and intracellular ROS production
Smad3, RhoA-GTP and intracellular ROS have been implicated in the EMT process.14 We observed that after 30 min of exposure, S1P (10−6 M) increased Smad3 phosphorylation, and only the S1P3 antagonist CAY10444 alleviated Smad3 phosphorylation (figure 6A). In other experiments, A549 cells were stimulated with S1P for 72 h, and phospho-Smad3 was quantified. The S1P-induced Smad3 phosphorylation after 72 h was suppressed by CAY10444 and JTE013, but not by W146 (figure 6B). Furthermore, anti-TGF-β1 antibody partially suppressed S1P-induced Smad3 phosphorylation, suggesting an autocrine role for TGF-β1 (figure 6B). In a similar fashion, S1P activated RhoA-GTP following 30 min and 72 h of S1P exposure, and this was suppressed by CAY10444 and JTE013, but not by W146 (figure 6C,D).
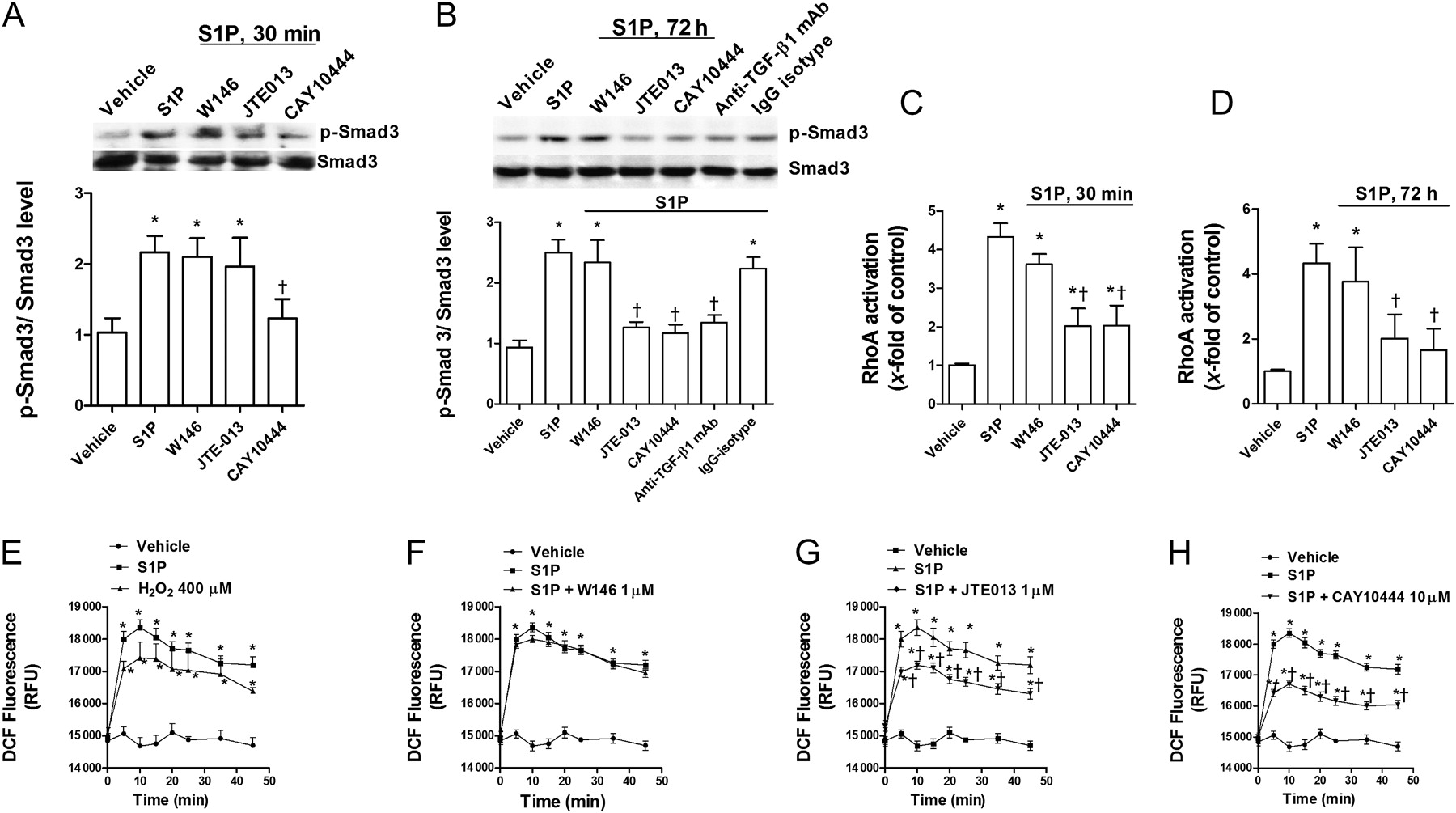
Sphingosine-1-phosphate (S1P) increases Smad3 phosphorylation, RhoA-GTP activation, and intracellular reactive oxygen species (ROS) generation. A549 cells were incubated with the S1P1 inhibitor W146 (1 μM), S1P2 inhibitor JTE013 (1 μM), S1P3 inhibitor CAY10444 (10 μM) or anti-transforming growth factor-β1 (TGF-β1) antibody (4 μg/ml) and its negative control (non-immune IgG) for 30 min before S1P (1 μM) stimulation for 30 min or 72 h. (A) Representative images of three western blots with respective densitometry analyses are shown for p-Smad3 and total Smad3 after 30 min of S1P exposure. (B) Representative images of three western blots with respective densitometry analyses are shown for p-Smad3 and total Smad3 after 72 h of S1P exposure. Results were corrected to total Smad3 and normalised to the non-treated vehicle group. Cell lysates were obtained after (C) 30 min or (D) 72 h of S1P exposure, and an ELISA-based RhoA-GTP activity assay was performed. (E, F, G and H) Intracellular ROS were determined by means of dichlorofluorescin (DCF) fluorescence intensity after S1P (1 μM) stimulation in the presence or absence of (F) W146 (1 μM), (G) JTE013 (1 μM) or (H) CAY10444 (10 μM). (E) H2O2 (400 μM) was used as a positive intracellular ROS inducer. Results are the means (SE) of three independent experiments. Post hoc Bonferroni tests: exact p values down to 0.05 are indicated in the graph (related to the indicated values or to solvent controls if placed on top of a bar); *p<0.05 related to solvent controls; †p<0.05 related to S1P-stimulated cell values as indicated.
S1P elicited an increase in intracellular ROS in a similar way as the positive control H2O2, with the highest levels after 10 min of stimulation. Pre-treatment with CAY10444 or JTE013 attenuated the S1P-induced ROS generation; pre-treatment with W146 was without effect (figure 6C).
S1P increases TGF-β1 expression and secretion
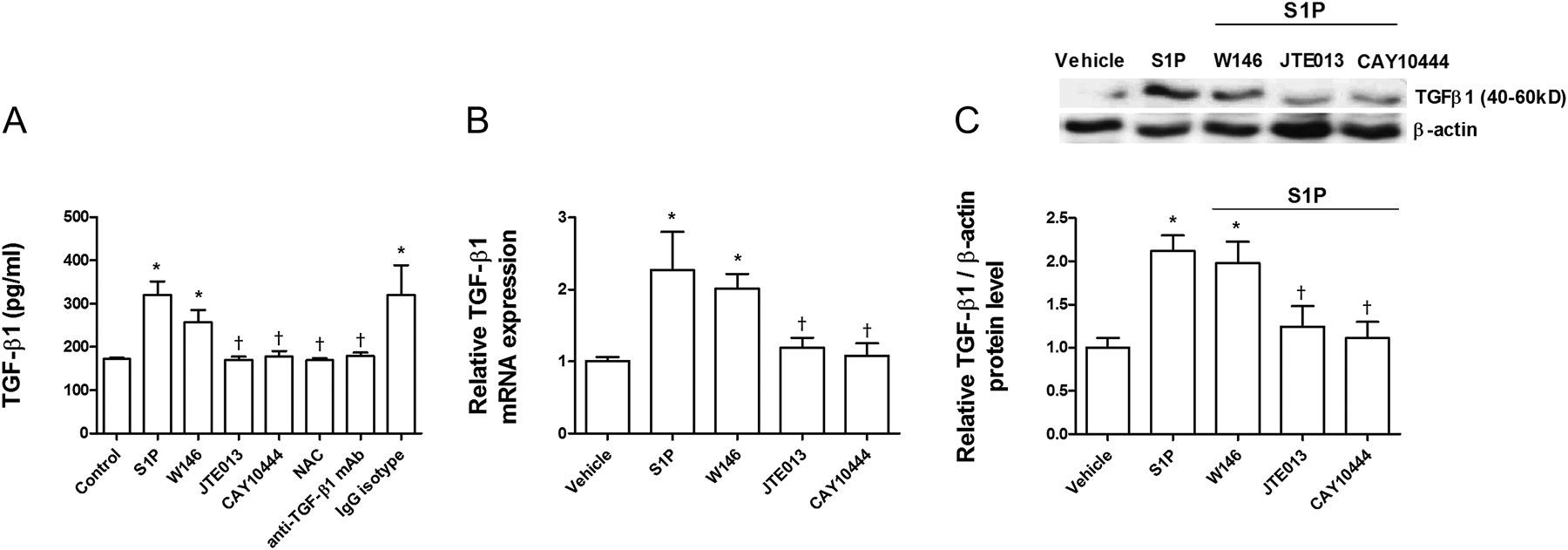
It was recently shown that intracellular ROS may induce TGF-β1 secretion.15 Because S1P promotes intracellular ROS formation, we explored whether S1P could also increase TGF-β1 expression and secretion. After 72 h of exposure, S1P (10−7 M) had increased TGF-β1 expression and secretion approximately twofold (figure 7A–C). JTE013 and CAY10444 attenuated S1P-induced TGF-β1 expression and secretion, whereas W146 had no effect (figure 7A–C). When intracellular ROS was removed by NAC, TGF-β1 remained at levels similar to control levels, suggesting a direct role of ROS in S1P-induced TGF-β1 expression. Moreover, anti-TGF-β1 antibody totally suppressed S1P-induced TGF-β1 expression, which confirms the removal capacity of this antibody.
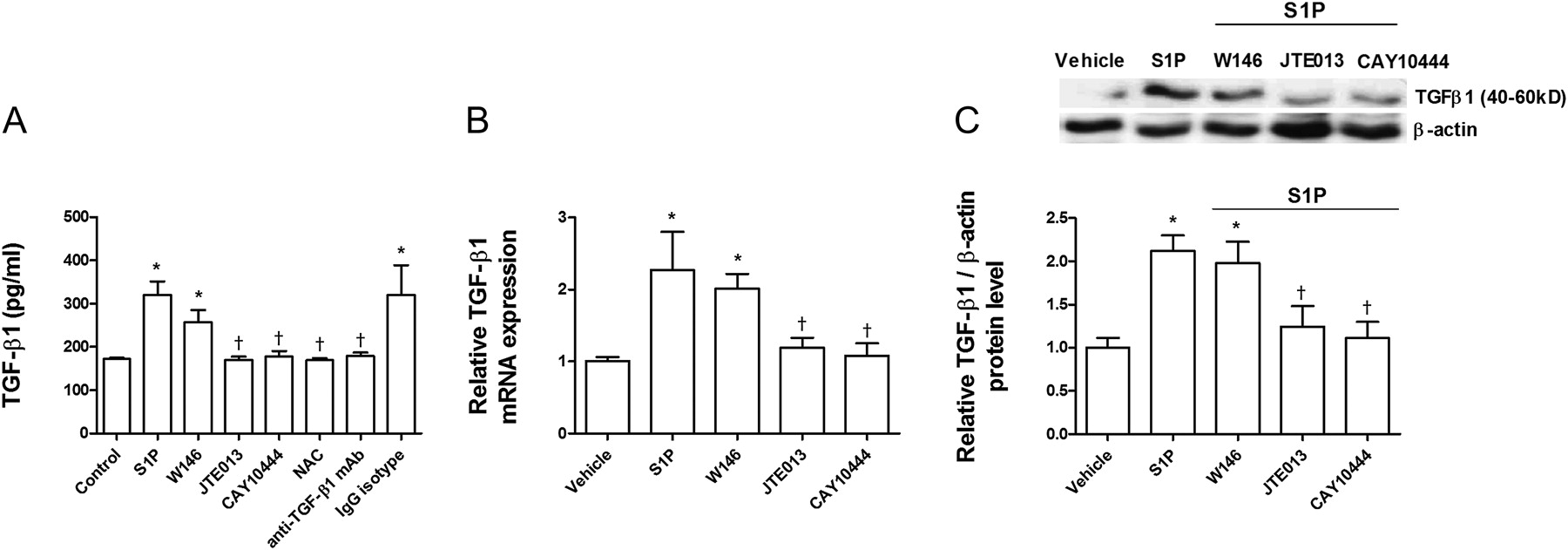
Sphingosine-1-phosphate (S1P) induces transforming growth factor-β1 (TGF-β1) expression and secretion. A549 cells were incubated with the S1P1 inhibitor W146 (1 μM), S1P2 inhibitor JTE013 (1 μM), S1P3 inhibitor CAY10444 (10 μM), antioxidant N-acetyl-L-cysteine (1 mM; NAC), or anti-TGF-β1 antibody (4 μg/ml) and its negative control (non-immune IgG) for 30 min before S1P (1 μM) stimulation for 72 h. (A) S1P increased TGF-β1 secretion as measured by ELISA. (B) S1P increased TGF-β1 mRNA and (C) protein expression as measured by real-time PCR and western blot analysis, respectively. (C) Representative images of three western blots with respective densitometry analyses are shown for TGF-β1 and β-actin as an internal control. Results are the means (SE) of three independent experiments. Post hoc Bonferroni tests: exact p values down to 0.05 are indicated in the graph (related to the indicated values or to solvent controls if placed on top of a bar); *p<0.05 related to solvent controls; †p<0.05 related to S1P-stimulated cell values as indicated.
TGF-β1-induced EMT is partially mediated by the SPHK1/S1P2–3 axis
TGF-β1 dose-dependently upregulated SPHK1 protein expression in A549 cells (figure 8A). However, we did not detect S1P in cell supernatants following TGF-β1 stimulation, probably because S1P is rapidly degraded by extracellular lipid phosphate phosphatases (data not shown).16

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Transforming growth factor-β1 (TGF-β1)-induced alveolar epithelial to mesenchymal transition (EMT) is partially mediated by the sphingosine-1-phosphate (S1P)/sphingosine kinase 1 (SPHK1) axis. (A) A549 cells were stimulated with TGF-β1 at different concentrations for 72 h, and SPHK1 expression was measured by western blot analysis. Representative images of three western blots with their respective densitometry analyses are shown for SPHK1 and β-actin as an internal control. (B) and (C) A549 cells were incubated with the Rho-kinase inhibitor Y27632 (10 μM), Smad3 inhibitor SIS3 (10 μM), antioxidant N-acetyl-L-cysteine (NAC; 1 mM), S1P1 inhibitor W146 (1 μM), S1P2 inhibitor JTE013 (1 μM), S1P3 inhibitor CAY10444 (10 μM) or SPHK inhibitor N,N-dimethylsphingosine (DMS) for 30 min before TGF-β1 (5 ng/ml, 72 h) stimulation. Following the incubation periods, mRNA was extracted, and the mesenchymal markers α-smooth muscle actin (α-SMA), vimentin and collagen (col) type I and epithelial markers E-cadherin and ZO-1 were quantified by real-time PCR. Results are the means (SE) of three independent experiments. Post hoc Bonferroni tests: exact p values down to 0.05 are indicated in the graph (related to the indicated values or to solvent controls if placed on top of a bar); *p<0.05; **p<0.01; ***p<0.001 related to solvent controls; †p<0.05; ††p<0.01 related to TGF-β1-stimulated cell values as indicated.
In other experiments, TGF-β1 (5 ng/ml, 72 h) induced EMT in A549 cells, as evidenced by increased α-SMA, vimentin and col type I mesenchymal markers and decreased E-cadherin and ZO-1 epithelial markers (figure 8B,C). Either JTE013 or CAY10444 as well as the SPHK inhibitor DMS attenuated TGF-β1-induced EMT. Furthermore, Y27632, SIS3 and NAC partially suppressed TGF-β1-induced EMT, confirming the participation of Rho kinase, Smad3 and ROS as previously mentioned.14
Discussion
The main and novel results of the present study are that serum S1P levels were upregulated in patients with IPF; S1P levels in BAL fluid were increased in patients with IPF and were inversely correlated with DLco, FEV1 and FVC; SPHK1 mRNA expression was upregulated in alveolar macrophages and lung tissue from patients with IPF and was correlated with the fibroblast markers α-SMA, vimentin and col type I; and S1P induced alveolar EMT in ATII and A549 cells through the activation of S1P2 and S1P3 with downstream signalling involving phospho-Smad3, RhoA-GTP and intracellular ROS, and by crosstalk between S1P/SPHK1 and TGF-β1 pathways. These new findings suggest that S1P may play a role in IPF, potentially by modulating the differentiation of alveolar epithelial cells to the mesenchymal phenotype as well as by the previously reported differentiation of fibroblasts into myofibroblasts.4 17
In the present work, serum S1P levels in the control group were 1 μM, which is consistent with previous data in normal subjects.18 In contrast, patients with IPF showed significantly higher serum S1P levels that reached 1.4 μM. The significance of these findings is currently unknown; however, based on previous results in animal models, we hypothesise that chronically elevated S1P levels disrupt endothelial barrier integrity and thereby contribute to lung fibrosis, as suggested by the exacerbated vascular leakage, fibrosis and mortality observed in mice exposed to the S1P analogue FTY720 after lung injury.19
A recent animal model of bleomycin-induced pulmonary fibrosis revealed that acid sphingomyelinase (ASMase) and acid ceramidase (AC) activities are increased in the lungs of fibrotic mice, and that ASMase knockout attenuated lung fibrosis.6 ASMase is a lysosomal enzyme responsible for the hydrolysis of sphingomyelin, which results in the production of ceramide and finally S1P. Furthermore, AC degrades ceramide to sphingosine, which is converted into S1P by means of SPHK. Bleomycin-induced ASMase and AC activation increased S1P levels in fibroblast cell cultures, which may explain the downstream effects of ASMase activation in bleomycin-induced mouse lung fibrosis.6 In support of these findings, it was recently shown that genetic deletion of SPHK1 protected mice from bleomycin-induced lung fibrosis and mortality.20 In the present study, the increase in SPHK1 expression in lung tissues from patients with IPF was correlated with α-SMA, vimentin and col type I expression. These results are in agreement with previous data showing upregulation of SPHK1, α-SMA, fibronectin and collagen in a mouse model of bleomycin-induced lung fibrosis.20 Interestingly, in SphK1−/− mice, the bleomycin-induced enhanced expression of α-SMA, fibronectin and collagen was attenuated, suggesting a potential role for SPHK1 in lung fibrosis.20 Along this line, it has been shown that SPHK1 and α-SMA are co-localised within mouse lung fibroblasts, confirming a common pattern of expression during lung fibrosis development.4 In this work, we observed increased SPHK1 expression located in fibroblasts and on the hyperplastic alveolar cells of patients with IPF, which represents a previously unappreciated finding and suggests a potential role for S1P in ATII and fibroblast activation and transformation. Besides the results observed in this study, some important limitations need to be discussed (see online supplementary data).
Following our evolving hypothesis, which suggests that IPF is a consequence of impaired wound healing involving the epithelial/fibroblast pathway, many candidate growth factor genes as well as other molecular mediators implicated in this extensive process have been evaluated.21 For example, TGF-β1 is a well characterised in vivo and in vitro growth factor mediating fibroblast and alveolar epithelial transformation into myofibroblasts.14 Myofibroblasts localise to fibrotic foci and other sites of active fibrosis and are the primary cell type responsible for the synthesis and deposition of ECM and the resultant structural remodelling that leads to the loss of alveolar function. SPHK1 inhibition suppresses the TGF-β1-induced fibroblast to myofibroblast transition,4 and previous reports have proposed a crosstalk between the S1P/SPHK and TGF-β1 pathways.13 Furthermore, TGF-β1 may trans-activate fibroblast S1P2 and S1P3, but not S1P1, to promote myofibroblast differentiation.4 13
Based on this background, we hypothesise that S1P may also promote alveolar EMT, which is one of the main processes of myofibroblast formation in IPF. In a recent study, S1P induced EMT in retinal pigmented cells.22 However, no data are available concerning alveolar epithelial cells. We observed that S1P induces alveolar EMT as assessed by increases in the myofibroblast markers α-SMA, vimentin and col type I and by the downregulation of the epithelial markers E-cadherin and ZO-1. The change in the ATII epithelial phenotype was accompanied by an arrest of cell growth. Although we previously reported that S1P promotes proliferation in A549 cells after 24 h,10 chronic stimulation during 72 h inhibited A549 cell growth. It is known that while cells are undergoing a differentiation process, they are not proliferative. Thus, for example, TGF-β1 inhibits A549 proliferation,23 and chronic S1P stimulation inhibits human keratinocyte proliferation.24
S1P-induced EMT was mediated by the activation of S1P2 and S1P3, which is in accordance with previous results in lung fibroblasts.4 However, S1P2 and S1P3 played a different role in the S1P-induced EMT process. S1P, through its interaction with S1P3, directly increased the phosphorylation of Smad3. In contrast, the S1P2 antagonist suppressed phospho-Smad3 expression following 72 h of S1P exposure, in a manner similar to that of the anti-TGF-β1 antibody. This observation may be explained because the S1P2 antagonist reduced S1P-induced TGF-β1 secretion after 72 h and therefore could indirectly inhibit Smad3 phosphorylation induced by TGF-β1, suggesting an autocrine role for TGF-β1 in the S1P-induced EMT process. This hypothesis was confirmed when the anti-TGF-β1 antibody attenuated the S1P-induced EMT process.
In addition, S1P-induced EMT was mediated by an increase in intracellular ROS and RhoA-GTP. Oxidative stress has been shown to promote TGF-β1 release.15 Thus, S1P-induced ROS could be responsible for TGF-β1 release and thereby for the EMT process. In the present study, the antioxidant NAC attenuated S1P-induced EMT as well as S1P-induced TGF-β1 release, confirming the crosstalk between S1P and TGF-β1. To further elucidate potential crosstalk mechanisms, we demonstrated that TGF-β1 exposure upregulated SPHK1, as previously reported in fibroblasts.4 Furthermore, TGF-β1-induced EMT in A549 cells was inhibited by an antagonist of SPHK as well as by antagonists of S1P2 and S1P3, providing more evidence for crosstalk between S1P/SPHK and TGF-β1. It is interesting to note that all of these intracellular pathways are related to IPF because ROS, Smad3 and RhoA-GTP are elevated in fibrosis and in the S1P-induced fibroblast-to-myofibroblast transition.4 25
In summary, this is the first report that identifies S1P as a potential marker of human IPF and inducer of alveolar EMT, which is known to participate in the progression of IPF. The effect of S1P on EMT could be explained by a crosstalk mechanism between the S1P/SPHK1 and TGF-β1 pathways. The data presented in this study suggest that S1P may be involved in the disease process and therefore future anti-fibrotic therapeutic interventions focused on the S1P system could be of potential value.
Acknowledgments
We are grateful for the valuable help of Professor M Guijarro and Professor Martorell of the Thoracic Surgery and Pathology Departments of the Valencia University General Hospital for providing lung tissue.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data Supplement - Manuscript file of format pdf
Footnotes
Funding This work was supported by grants SAF2008-03113 (JC), SAF2009-08913 (EJM), CIBERES (CB06/06/0027) from the Ministry of Science and Innovation and the Health Institute ‘Carlos III’ of the Spanish government, and research grants (Prometeo/2008/045 and Emerging Groups GE-029/10) from the regional government (‘Generalitat Valenciana’) and the Valencian Society of Pneumology.
Competing interests None.
Patient consent Obtained.
Ethics approval This study has been approved by the ethics committee of the University General Hospital of Valencia, Spain.
Provenance and peer review Not commissioned; externally peer-reviewed.
Linked Articles
- Airwaves