Article Text

Abstract
Background Bronchiolisation of distal airspaces is an unexplained feature of idiopathic pulmonary fibrosis (IPF). The authors sought to identify mechanisms driving the differentiation of mucus cells during the bronchiolisation process.
Methods Pathways governing airway mucus cell differentiation include SRY (sex determining region Y)-box 2 (SOX2), Notch, forkhead box A3(FOXA3)/SAM pointed domain containing ETS transcription factor (SPDEF), epidermal growth factor (EGF) and the EGF-related neuregulins NRG1α and NRG1β. Immunostaining for components of those pathways and mucins were performed on lung tissue obtained from patients with IPF (n=20), chronic obstructive pulmonary disease (n=13), idiopathic pulmonary artery hypertension (n=5) and from organ donors (n=6). NRG1α and NRG1β were quantified in bronchoalveolar lavage fluid (BALF) of patients with early IPF (n=20), controls (n=9), and patients with other interstitial pneumonias (n=13).
Results In IPF, the bronchiolised and enlarged distal airspaces stained for SOX2 are consistent with epithelial differentiation characteristic of conducting airway epithelium. IPF mucus cells expressed MUC5B but low levels of MUC5AC and MUC2, a profile typical of submucosal glands. Singularly, SPDEF, a transcription factor associated with mucus metaplasia, was rarely detected in mucus cells in IPF. The Notch target, HES1, was present in mucus cells from all groups. NRG1α was detected in serous cells within normal submucosal glands and in epithelial cells lining honeycombing areas in IPF, and was not detected in other patients. NRG1α concentrations were elevated in BALF from patients with early IPF.
Conclusion Expression of SOX2 and MUC5B and lack of SPDEF in atypically differentiated cells of bronchiolised distal airspaces are consistent with abnormal programming of airway epithelial cells in IPF. NRG1α may contribute to bronchiolisation of the distal lung seen in IPF.
Statistics from Altmetric.com
Key messages
What is the key question?
Abnormal differentiation of the respiratory epithelium is a feature of idiopathic pulmonary fibrosis (IPF). In this work, pathways governing mucus cell differentiation were investigated in lung tissues from patients with IPF and other chronic pulmonary disorders.
What is the bottom line?
Mucus cells lining abnormal airways from patients with IPF expressed MUC5B and SOX2 but lacked SAM pointed domain containing ETS transcription factor (SPDEF) and forkhead box A3(FOXA3). Neuregulin1α, which drives mucus cell differentiation in vitro, was expressed in normal airway submucosal glands and in lungs from patients with IPF.
Why read on?
Activation of abnormal respiratory epithelial differentiation programs may contribute to the expression of MUC5B and bronchiolisation of the distal lung, a salient feature of IPF.
In contrast to findings in chronic obstructive pulmonary disease and T helper 2-driven mucus metaplasia in which SPDEF, FOXA3, and MUC5AC are expressed at high levels, MUC5B was the predominant mucin and neither SPDEF nor FOXA3 were detected in the abnormal mucus cells characteristic of the IPF lesions.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a lethal disease affecting 6.8–16.3 per 100 000 individuals annually in the USA.1 While the mechanisms resulting in the loss of lung structure and function in this disorder remain poorly understood, there is increasing evidence that chronic injury to the respiratory epithelium activates aberrant repair processes, resulting in replacement of peripheral lung tissue with fibrotic scars and cysts lined by abnormal epithelial cells. These concepts are supported by experimental models in which epithelial cell injury2 or direct activation of remodelling processes leads to pulmonary fibrosis.3 Knowledge about the mechanisms influencing the differentiation and function of the respiratory epithelium in IPF may provide opportunities to diagnose, prevent or treat this incurable disorder.
Histopathological studies of IPF lungs reveal the typical ‘usual interstitial pneumonia’ pattern, with epithelial hyperplasia and apoptosis overlaying areas of scarring with fibroblast foci. In a focal manner, the alveolar architecture is destroyed, but regular lung structure may be found in adjacent regions. Bronchiolar abnormalities, including bronchiolisation of enlarged alveolar ducts, cysts and alveoli, are commonly seen, resulting in honeycombing when dilated airspaces communicate with proximal airways. Such bronchiolar changes are a salient feature of the disease.4 5 Recent data support the concept that bronchiolar abnormalities in IPF may be caused by cell autonomous changes in epithelial cell differentiation. Nuclear localisation of ß-catenin, indicating activation of the Wnt pathway, has been reported in bronchiolar-like epithelia lining the conducting airways in patients with IPF.6
An intriguing feature of the bronchiolar-like epithelium lining honeycombing regions in the IPF lung is the presence of mucin-laden cells, as mucus cells are normally absent from the distal lung.7 Moreover, while inflammation seems to be the main force inducing mucus cell differentiation in diseases such as asthma, cystic fibrosis, and chronic obstructive pulmonary disease (COPD), inflammation is usually mild in IPF.5 These discrepancies led us to hypothesise that the mechanisms governing mucus cell metaplasia in IPF may be distinct, and that their identification may provide insight into processes regulating cell fate and airway epithelial function in IPF. Mucus cells in the respiratory tract are thought to originate from the reversible differentiation of basal, secretory or ciliated epithelial cells. During development, activation of the Wnt/ß-catenin8 and Notch9 pathways increases mucus cell numbers in the airway. Postnatally, mucus cell differentiation of SOX2-expressing airway epithelial cells10 is strongly driven by the epidermal growth factor receptor (EGFR or ErbB1) and T helper 2 (Th2) cytokines, which activate the transcription factors forkhead box A3 (FOXA3) and SAM pointed domain containing ETS transcription factor (SPDEF) in association with downregulation of FOXA2.11 12 Recently, activation of other members of the ErbB family by neuregulin-1 (NRG1) splice variants NRG1α and NRG1β was shown to enhance transcription of mucin genes and to increase the number of mucus cells in vitro.13 14 In the gut, SPDEF,15 Notch/AtoH16 and Krüppel-like factor 4 (KLF-4)17 transcriptionally influence mucus cell differentiation. Airway mucus cell differentiation is also influenced by site-specific cues. While MUC5B is the predominant mucin produced in submucosal glands, mucus cells lining conducting airways generally express MUC2 and MUC5AC18 and do not express MUC5B at high levels.
In the present study, the mechanisms governing mucus cell differentiation were assessed in the lungs of patients with IPF, in patients free of lung disease and in patients with other chronic lung disorders. The expression of transcription factors, ligands and/or activation markers indicating activity of the SPDEF, Notch, Erb2/3/4, and KLF-4 pathways was determined.
Materials and methods
Lung samples
Anonymous lung samples were obtained at the University of Vienna and the University of Giessen Lung Center, at the time of lung transplantation from patients with IPF (n=20), COPD (n=13), and with idiopathic pulmonary arterial hypertension (IPAH, n=5); donor lobes resected because of size incompatibility served as controls (n=6). Bronchial samples were obtained during lung cancer resection surgery at the University Hospital in Cincinnati. Patient characteristics are reported in online table S1. IPF was diagnosed according to American Thoracic Society/European Respiratory Society (ATS/ERS) criteria.19 Utilisation of human lung samples was reviewed and approved by the Ethics Committee of Justus-Liebig-University of Giessen.
Immunohistochemistry
Five micrometre sections were deparaffinised and endogenous peroxidases were blocked. After antigen retrieval in 0.1 M citrate (pH 6) at 95°C, sections were blocked with non-specific sera and primary antibodies (online table S2) were incubated overnight at 4°C. Non-immune immunoglobins were used as negative controls. The HES120 and SPDEF21 antibodies were gifts from Dr Stanger (University of Pennsylvania) and Dr Watson (University of South Carolina). Biotinylated secondary antibodies and biotin-conjugated horseradish peroxidase complexes (both from Vector Laboratories, Burlingame, California, USA) were sequentially applied and staining was developed with nickel-diaminobenzidine (Sigma, St Louis, Missouri, USA). Staining by the NRG1α (RB277) antibody was completely blocked when this antibody was pre-incubated with recombinant human NRG1α EGF-like domain (296-HR, R&D Systems, Minneapolis, Minnesota, USA). Sections were counter-stained with Alcian Blue and Nuclear Fast Red (Poly Scientific, Bay Shore, New York, USA) and examined at ×100 magnification. Mucus cells were identified by Alcian Blue staining of the cytoplasm. The number of cells expressing MUC2, MUC5B, MUC5AC, FOXA3, SPDEF and HES1 was counted on each whole section and was expressed as a percentage of the total number of mucus cells on the section.
Dual labelling to identify NRG1α and p63
After NRG1α staining, sections were incubated with a mouse monoclonal antibody recognising p63 (sc-56188, Santa Cruz Biotechnology, Santa Cruz, California, USA), exposed to an alkaline phosphatase-conjugated goat anti-mouse antibody (31320, Thermo Scientific, Waltham, Massachusetts, USA), developed with Fast Red TR/Naphthol AS-MX (Sigma), and counterstained with haematoxylin.
Bronchoalveolar lavage fluid samples
Bronchoalveolar lavage fluid (BALF) was obtained at Bichat University Hospital (Paris, France) at the time of initial diagnostic evaluation in patients with IPF (n=20), in patients free of interstitial lung disease (n=9), and in patients with non-IPF chronic interstitial pneumonias (n=13). Patients gave informed consent and the protocol was approved by the ethics committee of Hôtel-Dieu Hospital (Paris, France). Patient characteristics are reported in online table S3. BALF was concentrated fivefold with Amicon Ultra 3K columns (Millipore, Billerica, Massachusetts, USA).
NRG1α and NRG1β ELISA
Plates were coated with a goat anti-NRG1α capture antibody (AF296, R&D Systems) and blocked with albumin. Standards were prepared with human NRG1α EGF-like domain. A rabbit anti-NRG1α detection antibody (RB277, Thermo Scientific) was applied followed by a peroxidase-conjugated anti-rabbit antibody (#401315, Calbiochem, Gibbstown, New Jersey, USA). Tetramethylbenzidine (Sigma) was used as a substrate. NRG1β levels were determined with a commercial ELISA (DY377, R&D Systems).
Statistical analysis
Comparisons between multiple groups were performed using Kruskall–Wallis' analysis of variance test followed by two-by-two comparisons with Mann–Whitney's U-test when a difference was detected. Immunohistochemical and BALF data were illustrated as box plots showing the 10th, 25th, 50th, 75th and 90th percentiles. Clinical data were presented as mean±standard error of the mean (SEM). Correlations between continuous variables were determined using Spearman's correlation test and linear regression. Associations between categorical variables were assessed using the χ2 test. A p value less than 0.05 was considered significant.
Results
Distinct morphology of the peripheral bronchiolar epithelium in IPF
In the lungs of organ donors and patients with COPD and IPAH, airways were lined by a pseudostratified epithelium in which mucus cells were interspersed with basal, ciliated, and serous cell types. Differentiated mucus cell morphology was readily observed. In sharp contrast, peripheral bronchioles in honeycombed areas in lungs from patients with IPF were lined by mucus cells that were generally of low columnar morphology and usually organised as a homogenous single-layered epithelium. Such morphological appearance was observed in 53.5% of patients, and was never observed in donor, COPD or IPAH lungs (p<0.0001, χ2 test).
Mucus cells lining bronchiolar lesions in IPF expressed SOX2
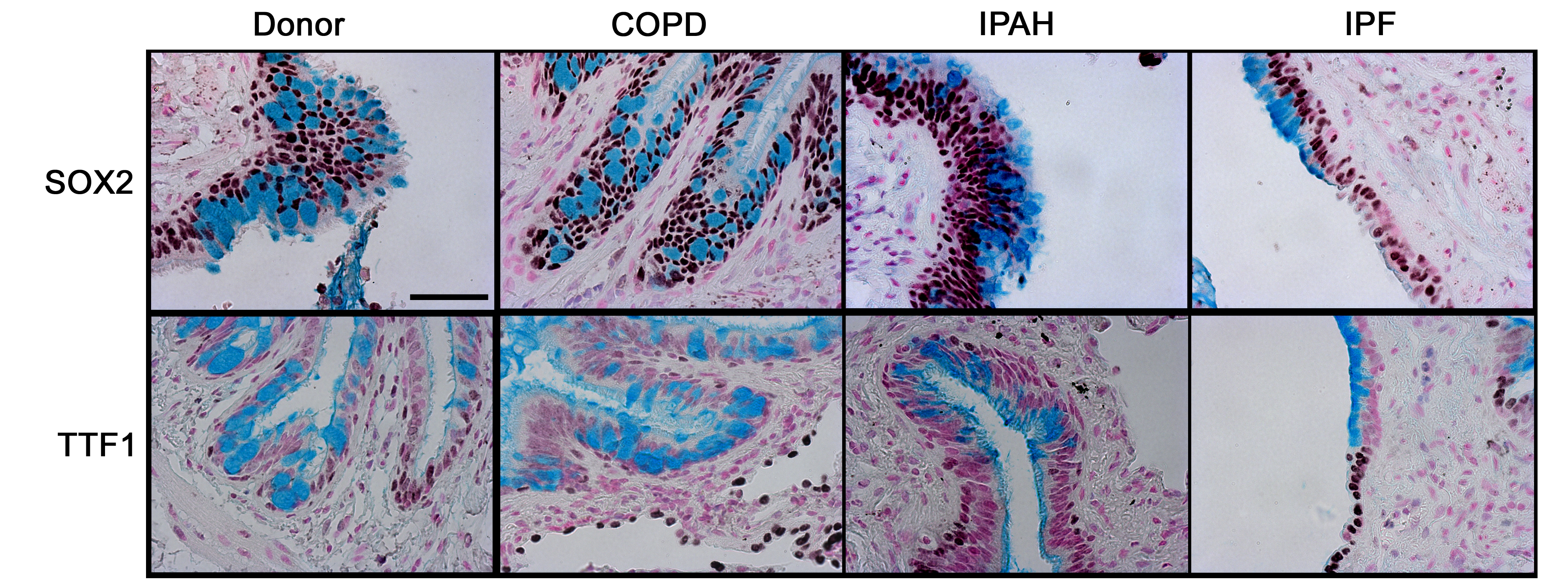
SOX2 is a transcription factor expressed selectively in conducting airway epithelial cells but not in alveolar regions.22 SOX2 drives proliferation and differentiation of the respiratory epithelium towards conducting airway epithelial cell types.23 24 SOX2 was detected in epithelial cells lining conducting airways in lungs from controls and patients with COPD, IPAH, and IPF (figure 1 and online figure s1). Mucus cells lining the honeycombed regions of lungs from patients with IPF were uniformly stained by the antibody against SOX2, consistent with cells lining conducting airways, but not alveolar regions. TTF1, a transcription factor expressed strongly in alveolar type II cells and at lower levels in conducting airways, was neither detected in mucus cells from IPF nor from patients without IPF. In contrast to the lack of TTF1 in honeycombed regions, TTF1 was highly expressed in hyperplastic alveolar epithelial cells in patients with IPF.
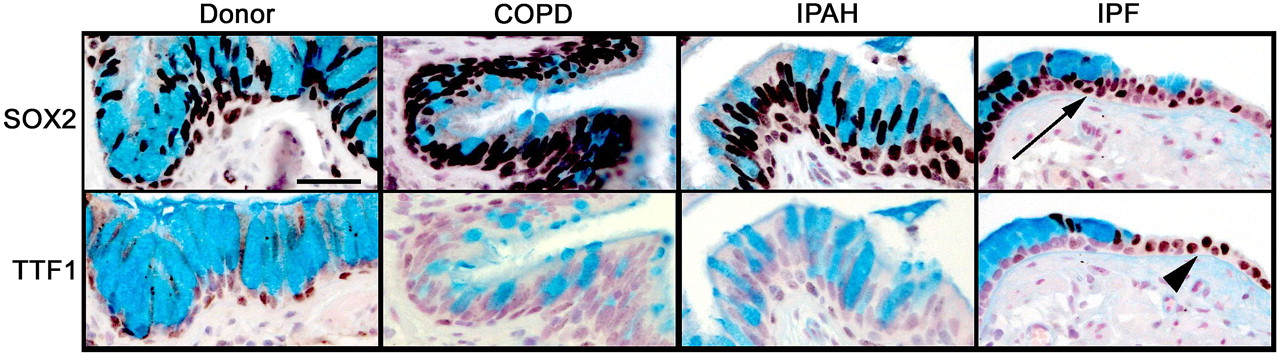
Alcian blue, SOX2, and TTF1 staining in the distal airway epithelium. Tissue sections from organ donors (n=7) and patients with chronic obstructive pulmonary disease (COPD) (n=13), idiopathic pulmonary arterial hypertension (IPAH) (n=5) and idiopathic pulmonary fibrosis (IPF) (n=20) were stained for SOX2 and TTF1 (brown/black colour), and counter-stained with Nuclear Fast Red and Alcian Blue, the latter staining mucus cells. Airway mucus cells expressed SOX2 (arrow) and lacked TTF1 which was expressed by neighbouring epithelial cells in the lung with IPF (arrowhead). Serial sections are shown. Scale bar: 30 μm.
Distinct mucin expression profile in IPF
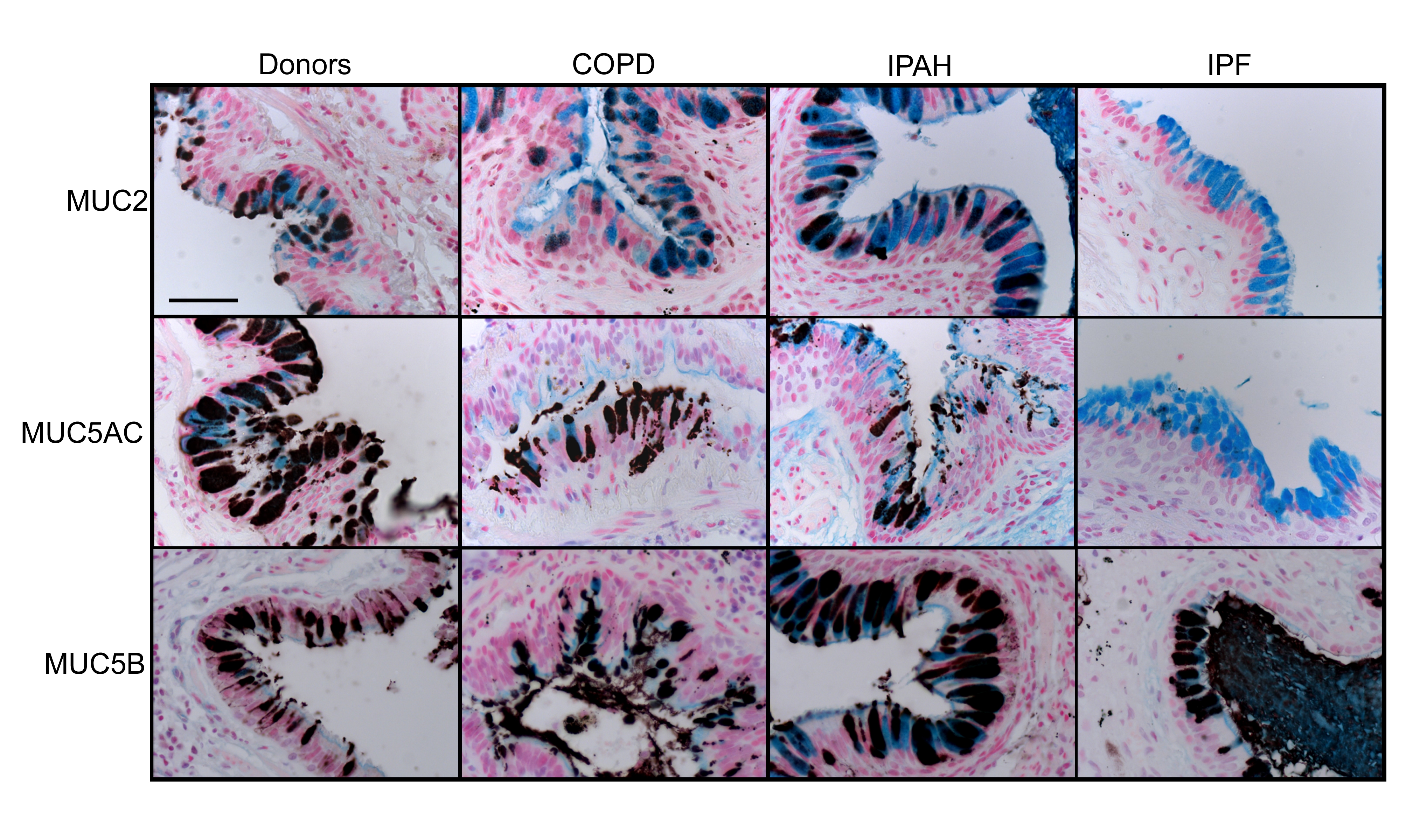
Mucus cells in lungs from donors and patients with COPD and IPAH expressed MUC2, MUC5AC, and MUC5B (figure 2 and online figure s2). In contrast, mucus cells lining IPF lesions expressed MUC5B, and rarely MUC2 or MUC5AC (p=0.018 and p=0.0014 in comparison to controls, respectively).

Distinct mucin expression profile in idiopathic pulmonary fibrosis (IPF). Sections from organ donors (n=7) and patients with chronic obstructive pulmonary disease (COPD) (n=13), idiopathic pulmonary arterial hypertension (IPAH) (n=5) and IPF (n=20) were immunostained for mucins (black colour) and counter stained with Nuclear Fast Red and Alcian Blue. MUC2, MUC5AC, and MUC5B immunostaining are shown in figure 2A. Scale bar: 30 μm. Serial sections are shown. The percentage of mucus cells expressing MUC2, MUC5AC and MUC5B in the lungs are shown in figure 2B. Box plots show 10th, 25th, 50th, 75th and 90th percentiles. *p<0.05 compared with donors.
Activation of the SPDEF differentiation pathway was lacking in IPF mucus cells
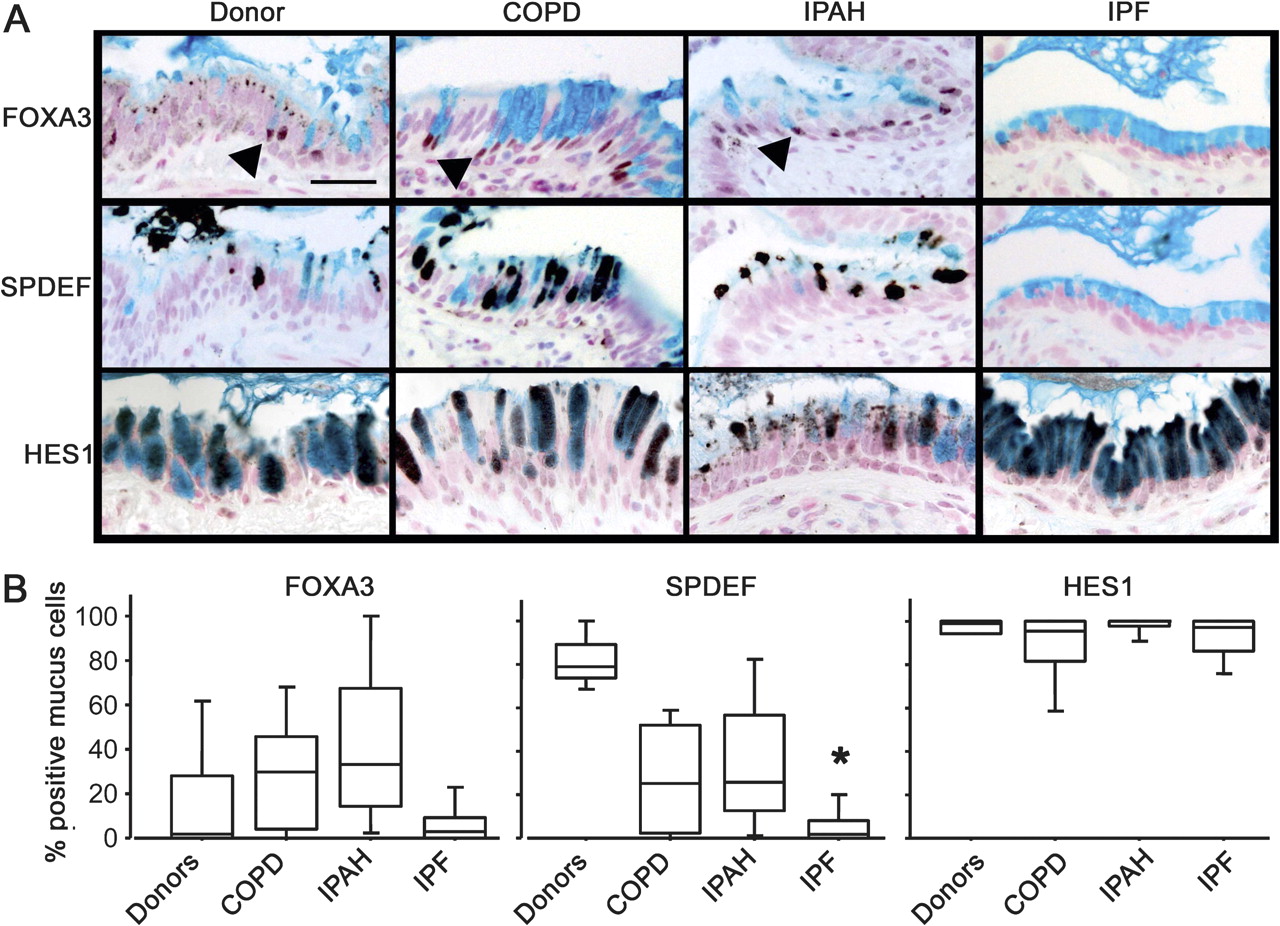
Mucus cell differentiation in adult airways is influenced by Th2 cytokine-driven mechanisms controlled by the transcription factor SPDEF. Recent studies demonstrated that SPDEF is both necessary and sufficient to induce mucus cell differentiation in mice, and is strongly expressed in the lungs of patients with cystic fibrosis or after chronic smoking.11 SPDEF activates MUC5AC and FOXA3 following Th2 cytokine activation. While the mucus cells in patients with COPD, IPAH, and controls frequently stained with antibodies against FOXA3 and particularly SPDEF (figure 3 and online figure s3), the mucus cells in patients with IPF rarely stained for SPDEF (p=0.0014 in comparison to controls) or FOXA3, indicating that mucus cell differentiation in IPF likely occurs by distinct processes. Supporting this hypothesis, the transcription factor FOXA2, which is down-regulated by SPDEF, was frequently detected in the nuclei of mucus cells of patients with IPF (not shown). Staining for SPDEF was correlated with the expression of MUC5AC in controls and patients with COPD and IPAH (Spearman's test: p=0.0047, figure 4).
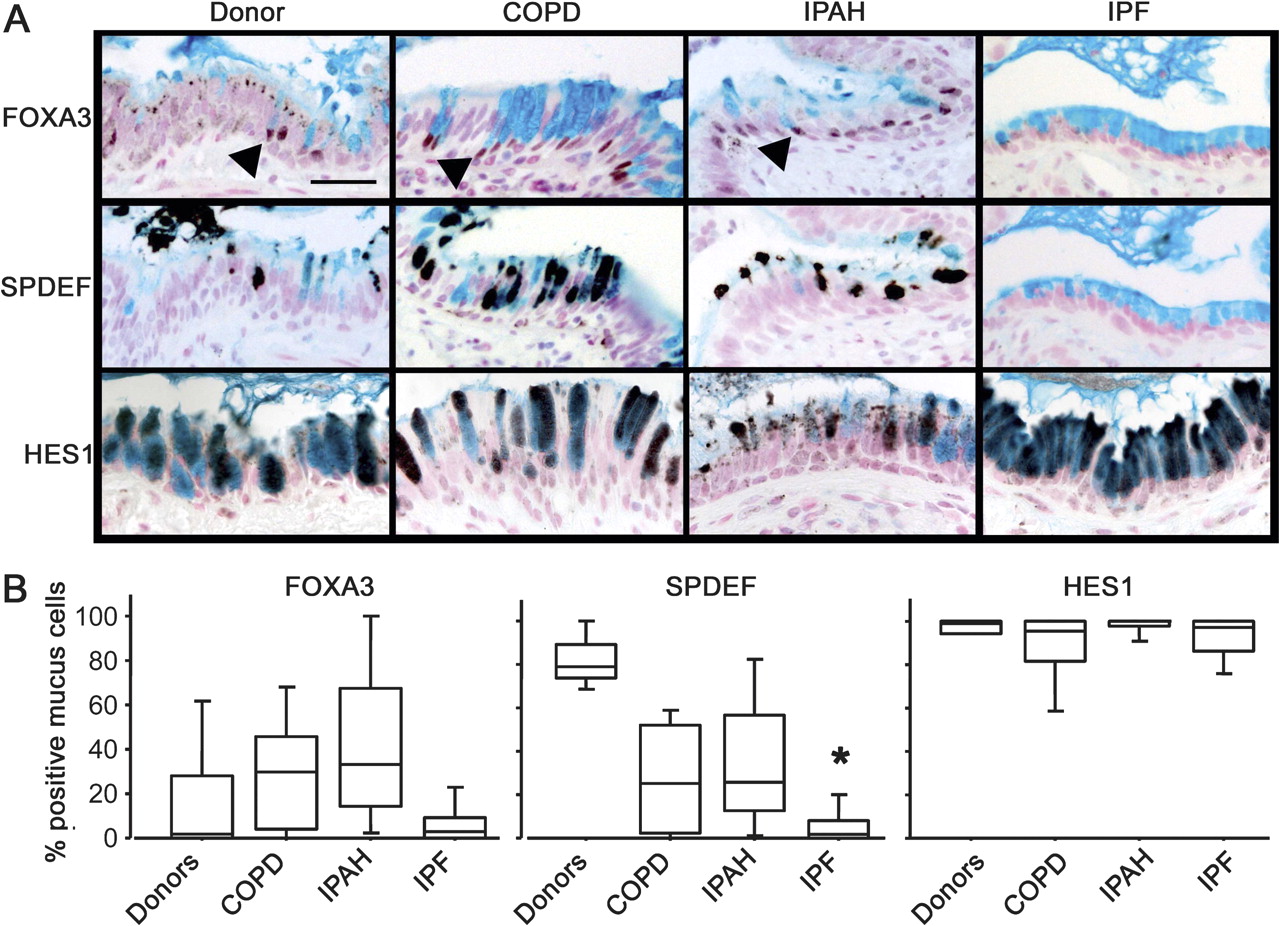
Lack of SPDEF and FOXA3 staining in idiopathic pulmonary fibrosis (IPF). Sections from organ donors (n=7) and patients with chronic obstructive pulmonary disease (COPD) (n=13), idiopathic pulmonary arterial hypertension (IPAH) (n=5) and IPF (n=20) were immunostained for FOXA3, SPDEF (serial sections), and HES1 (brown/black colour) and counter stained with Nuclear Fast red and Alcian blue as shown in figure 3A. Scale bar: 30 μm. Arrowheads show FOXA3-positive nuclei. The percentage of mucus cells expressing FOXA3, SPDEF and HES1 in the lungs is shown in figure 3B. Box plots show 10th, 25th, 50th, 75th and 90th percentiles. *p<0.05 compared with donors.
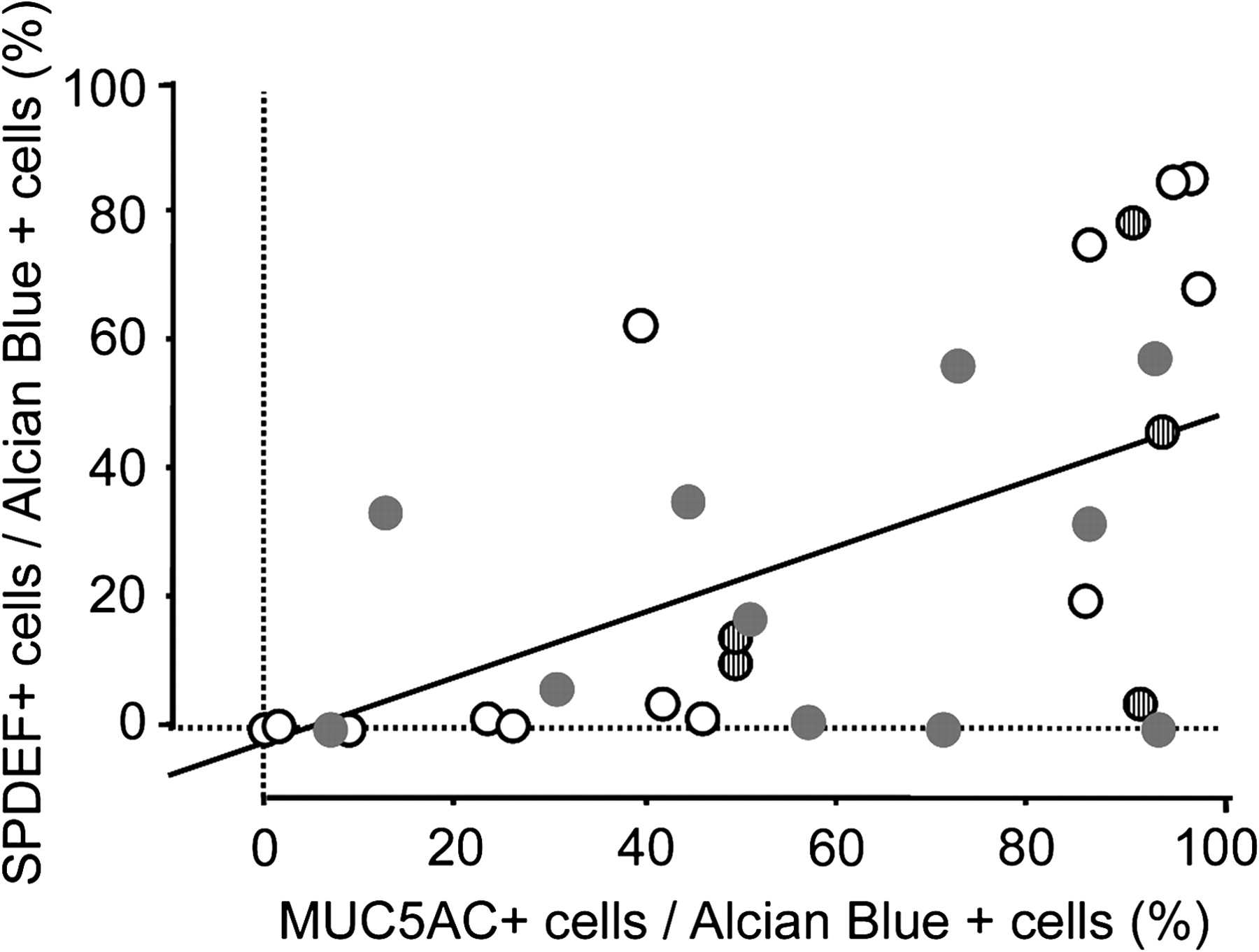
Correlation between MUC5AC and SPDEF. MUC5AC staining was correlated with that for SPDEF. Linear regression graph showing association between the percentage of mucus (Alcian blue positive) cells expressing SPDEF and MUC5AC in the lungs of organ donors (empty dots) and patients with chronic obstructive pulmonary disease (COPD) (grey dots), idiopathic pulmonary arterial hypertension (IPAH) (striped dots). p=0.0015, R2=0.291.
Mucus cells from all groups of patients were selectively stained by an antibody against HES1, indicating Notch pathway activation (figure 3). KLF-4, which is expressed at the mRNA level in the lungs of patients with rapidly progressive IPF,25 was not detected in lung tissue from any patient groups, although it was detected in mucus cells in human colonic epithelia (data not shown).
NRG1α was expressed in airway epithelial cells in IPF
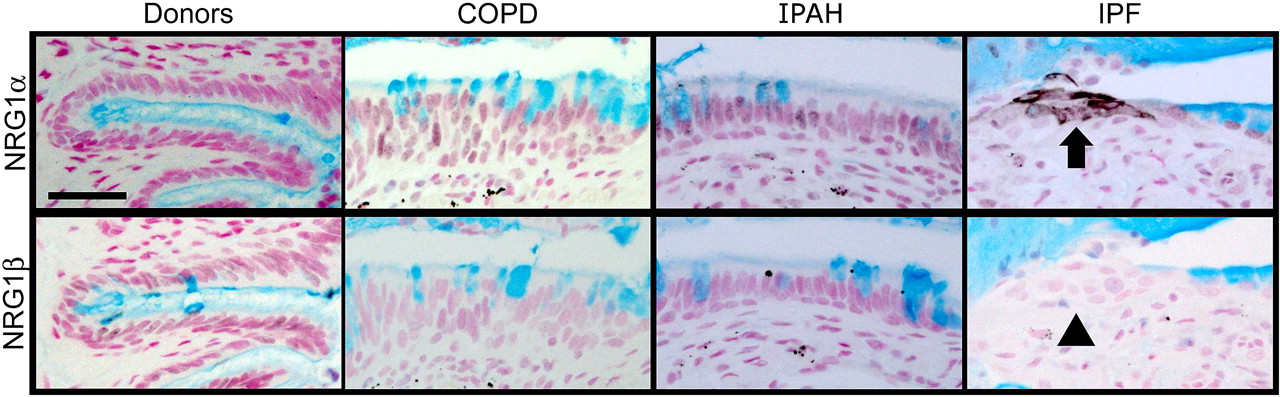
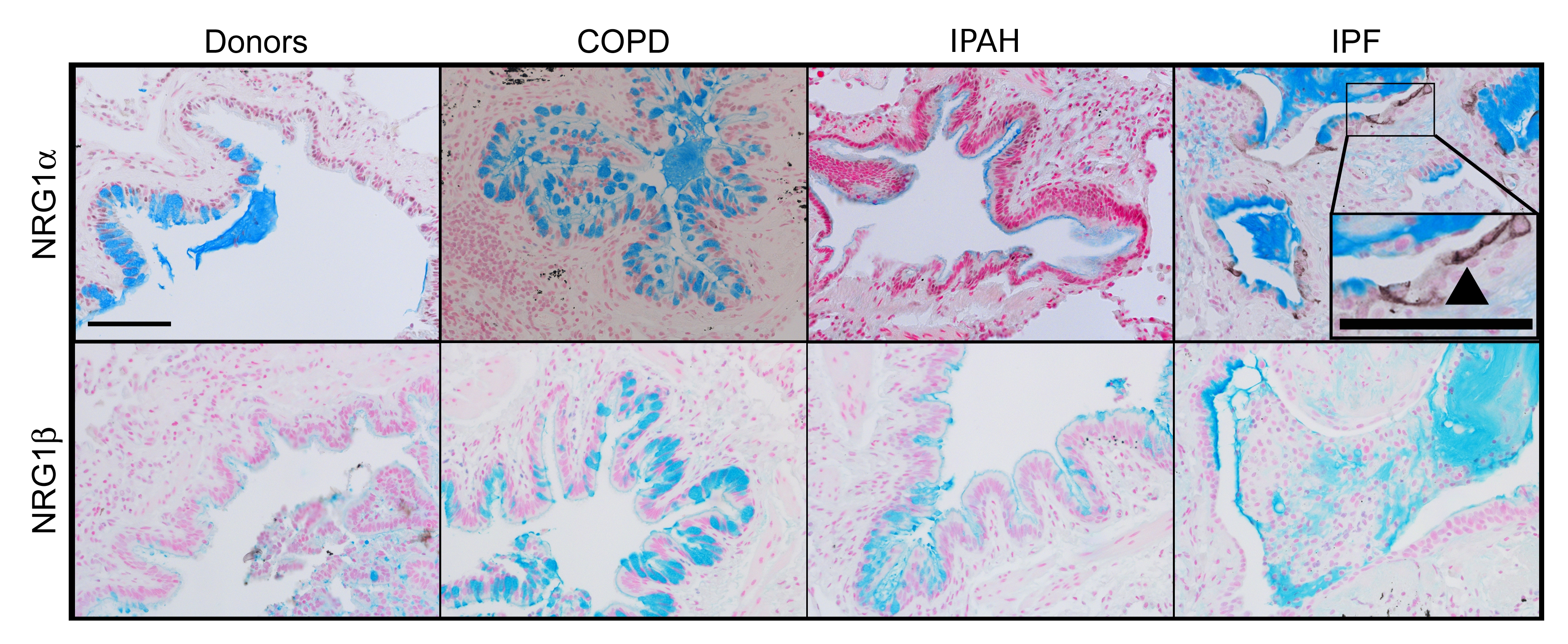
NRG1α and NRG1ß induce mucus cell differentiation in human airway epithelial cells in vitro.14 The mucin expression profile of cells treated with NRG1α, but not NRG1ß, is similar to those in the IPF samples currently studied. While NRG1α staining was rarely and sporadically detected in tissue from patients with COPD (15%) or IPAH (16%), NRG1α was readily detected in a majority of patients with IPF (56%, p=0.0002, χ2 test) where it was focally present in the cytoplasm and plasma membranes in a subset of epithelial cells lining bronchiolised regions (figure 5 and online figure s4). NRG1ß was not detected in normal submucosal glands or in airways or alveolar regions of tissues from either donors or patients with COPD, IPAH or IPF, but was readily detected in human tonsils (not shown).

NRG1α staining in idiopathic pulmonary fibrosis (IPF) lung tissue. Lung sections from organ donors (n=7) and patients with chronic obstructive pulmonary disease (COPD) (n=13), idiopathic pulmonary arterial hypertension (IPAH) (n=5) and IPF (n=20) were immunostained for NRG1α and NRG1β (black colour) and counter stained with Nuclear Fast Red and Alcian Blue. The mucus cell differentiation factor NRG1α was detected in a subset of airway epithelial cells in patients with IPF (arrow); these cells did not express NRG1β (arrowhead). Serial sections are shown. Scale bar: 30 μm.
Association of NRG1α with p63
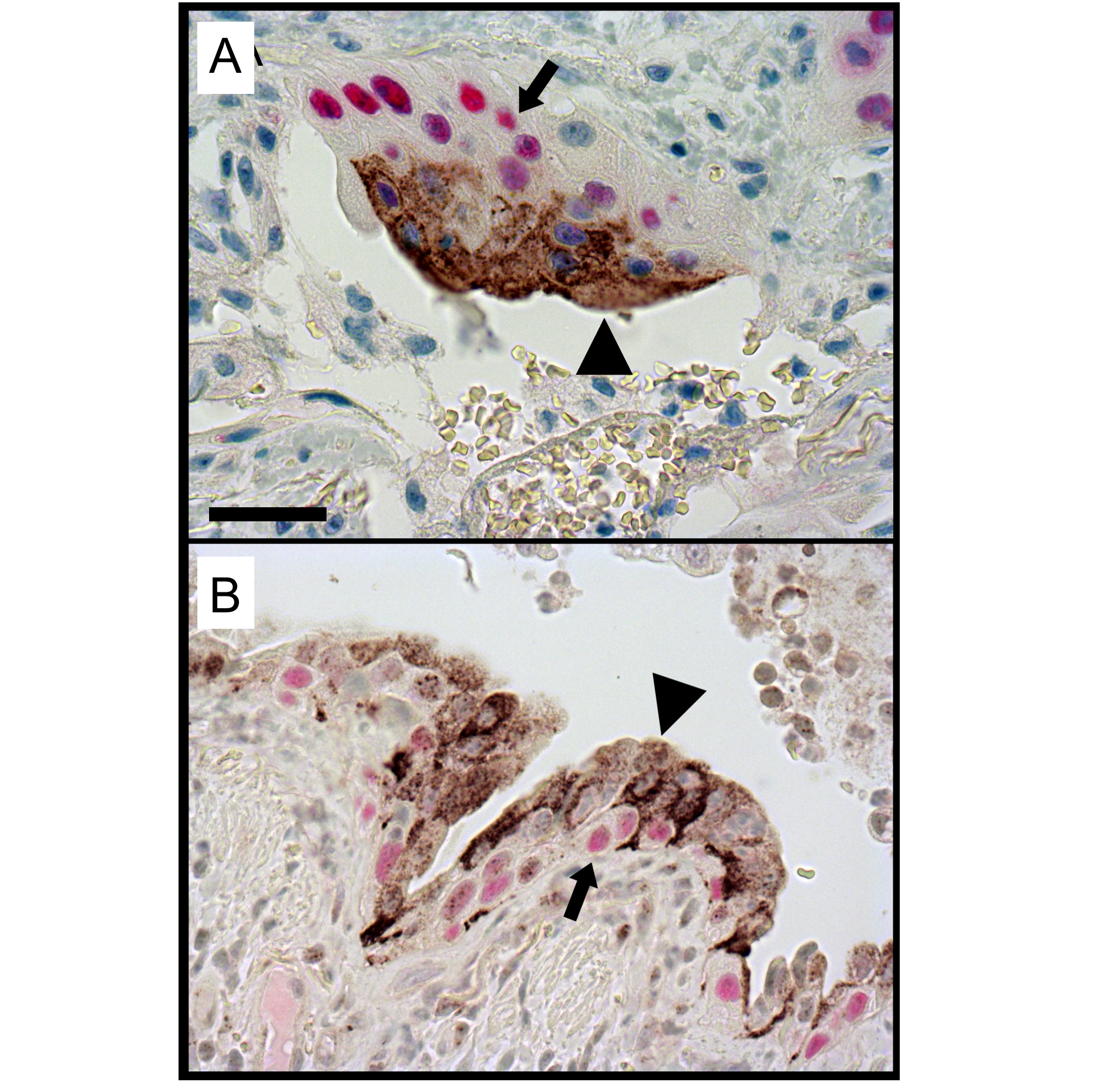
NRG1α-stained epithelial cells in the lungs of patients with IPF were generally squamous in morphology. Double immunofluoresence demonstrated that the NRG1α-positive cells were generally located in close proximity to p63-positive cells within pseudostratified/stratified regions of the bronchiolised epithelium in IPF (figure 6 and online figure s5).

Cellular localisation of NRG1α and p63 in the idiopathic pulmonary fibrosis (IPF) lesions. Sections from patients with IPF were co-immunostained for NRG1α (brown color) and p63 (red color) and counterstained with haematoxylin. Squamous NRG1α-positive cells (arrowheads) were localised in close apposition to p63-positive basal cells (arrows) in the distal lung of patients with IPF. Image is representative of n=12 experiments. Scale bar: 50 μm.
MUC5B and NRG1α were expressed in normal proximal airway submucosal glands
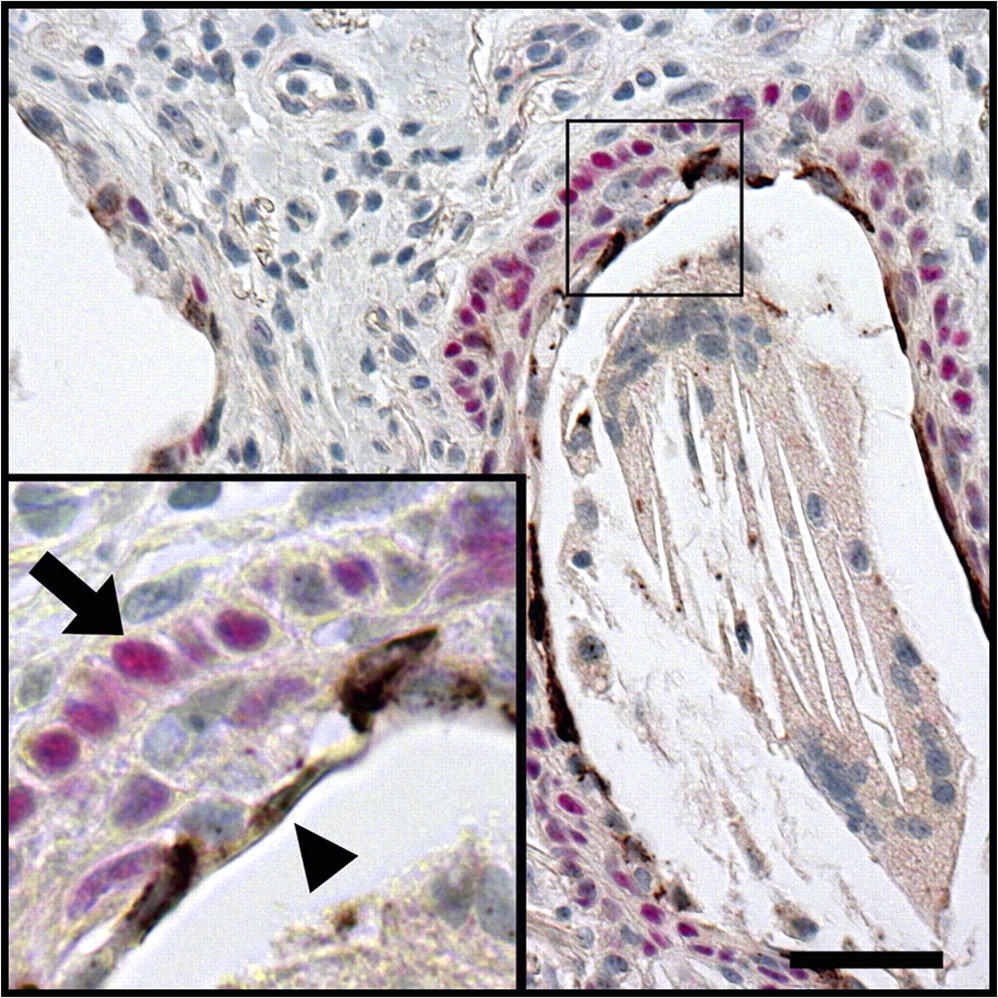
In striking similarity to the distal lung of patients with IPF, mucus cells in normal proximal airway submucosal glands expressed MUC5B but undetectable levels of MUC5AC, while NRG1α was detected in serous cells, in close proximity to p63-positive cells (figure 7).

Normal proximal airway submucosal glands share common features with the peripheral lung lesion in idiopathic pulmonary fibrosis (IPF) patients. Normal submucosal gland sections were immunostained for MUC5AC (black, figure 7A), MUC5B (black, figure 7B), and co-stained for NRG1α (brown) and p63 (red, figure 7C), with Nuclear Fast Red counterstaining. Normal proximal airway submucosal gland mucus cells expressed MUC5B but not MUC5AC. NRG1α-expressing serous cells (arrows) were apposed to p63-positive basal cells (arrowheads). Photomicrographs representative of two patients. Scale bar: 50 μm.
Increased levels of NRG1α in BALF in early-stage IPF
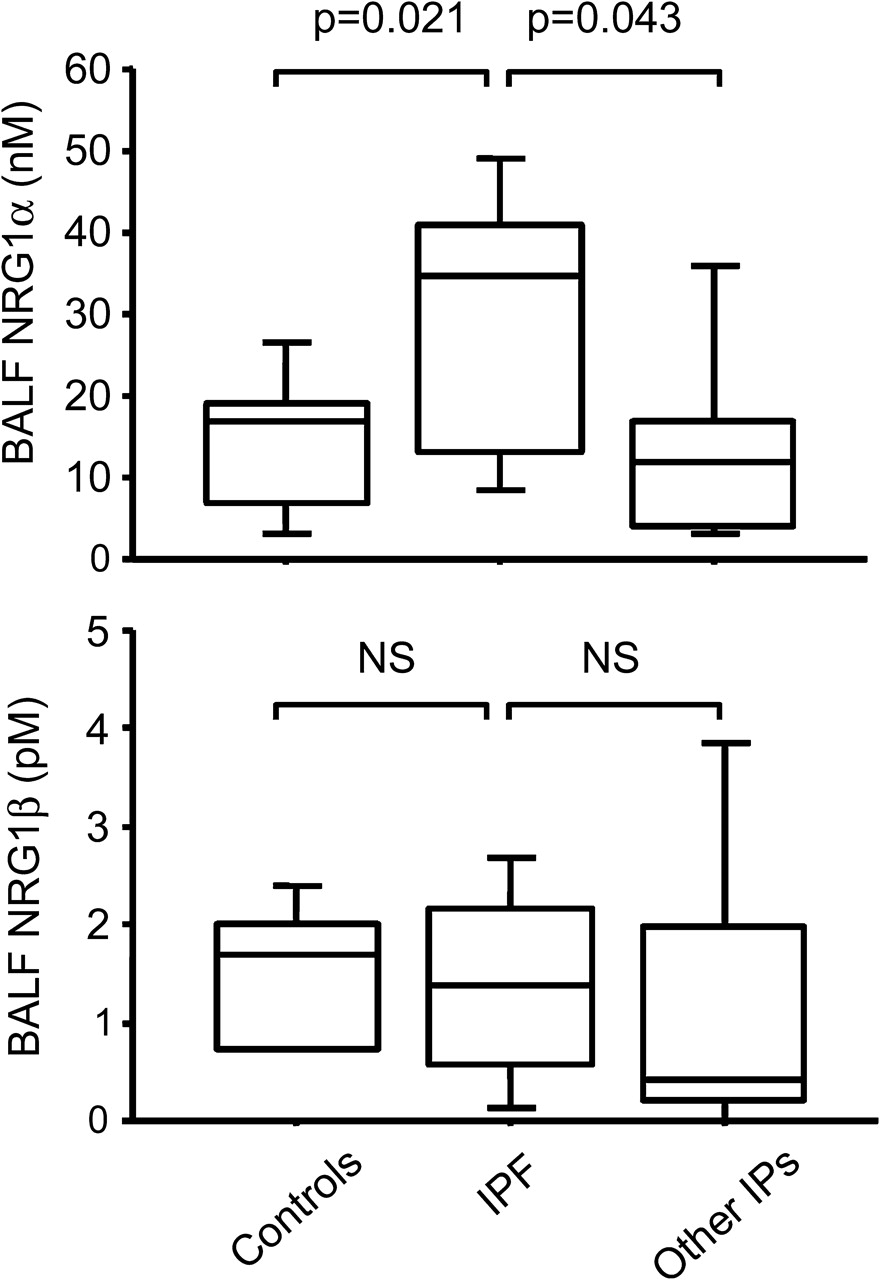
To determine whether increased epithelial NRG1α expression was an early event in IPF, we compared BALF NRG1α and NRG1β levels in patients with early IPF, in patients with other chronic interstitial lung disease and in controls. BALF NRG1α levels were higher in patients with IPF (29.2±4.6 nM) compared with controls (14.2±2.9 nM, p=0.021) and with patients with non-IPF chronic interstitial lung disease (21.6±7.9 nM, p=0.043). NRG1β levels were similar in all groups of patients (figure 8).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Quantitation of NRG1α in bronchoalveolar lavage fluid (BALF) in early stage idiopathic pulmonary fibrosis (IPF). Increased levels of NRG1α in BALF of patients with early-stage IPF. NRG1α and NRG1ß concentrations determined by ELISA in BALF of patients free of chronic lung disease, in patients with early IPF and in patients with other chronic interstitial pneumonias (IPs). Box plots show 10th, 25th, 50th, 75th and 90th percentiles.
Discussion
There is increasing evidence that chronic alveolar type II cell injury, whether related to endoplasmic reticulum26 or lysosomal27 stress, is involved in the pathogenesis of IPF. Recurrent epithelial cell death induces a regenerative response associated with activation of embryologically important pathways such as Wnt/β-catenin.6 Fibroblast foci develop progressively and a marked distortion of alveolar structure occurs. Septal structures are replaced by enlarged distal airspaces or cysts typically covered by epithelial cells with characteristics of proximal airway epithelium. Expression of SOX2 and paucity of TTF1 in epithelial cells in the bronchiolised distal airspaces in IPF support this concept.
In IPF, the airways were often lined by atypical, cuboidal mucus cells. In contrast to the abundant expression of SPDEF, MUC5AC and MUC2 by mucus cells in COPD and other chronic lung diseases, mucus cells in IPF expressed MUC5B, a mucin expressed at higher levels in submucosal glands, and lacked SPDEF, implicating the activation of specific cell differentiation pathways. NRG1α expression was detected in the bronchiolised distal airspaces in IPF as well as in normal airway submucosal glands, supporting the concept that NRG1α influences a mucus cell differentiation program distinct from that seen in the surface airway epithelium of patients with COPD or cystic fibrosis.11
In the airways of organ donors as well as patients with COPD and IPAH, expression of SPDEF by mucus cells was correlated with expression of MUC5AC, further supporting the concept of a causal association between SPDEF expression and mucus metaplasia. Interestingly, as seen in mouse models,11 12 SPDEF was prominent in the cytoplasm of mucus cells. It is unclear whether SPDEF functions solely as a nuclear transcription factor or influences mucus cell differentiation via mechanisms that do not require direct activation of target genes.12 FOXA3 was infrequently detected in mucus cells of patients with and without IPF. In line with the finding that FOXA3 expression is not associated with airway mucus metaplasia in patients with asthma,28 this result suggests that FOXA3 may not play a critical role in mucus metaplasia in humans.
NRG1α, which enhances MUC5B but not MUC5AC expression in human airway epithelial cells, was expressed in the lungs of 56% of patients with IPF, being localised in squamous epithelial cells located in close proximity to basal cells expressing p63 (figure 6). NRG1α may also be expressed by other squamous epithelia. The lack of NRG1α detection in the remaining patients may have been related either to absent NRG1α expression or, as NRG1α was focally expressed in IPF lungs, to sampling of areas of the lung not expressing NRG1α. NRG1α belongs to a family of signalling proteins that exert their effects through the EGFR-related receptors ErbB2, ErbB3 and ErbB4. NRG1 is a large and complex gene from which multiple protein isoforms are produced using several promoters and splicing; α and β isoforms of NRG1 differ in their EGF-like domain. While the NRG1 gene is necessary for heart and brain development,29 mice carrying a deletion of NRG1α EGF-like domain develop normally.30
The fact that high levels of NRG1α were detected in the BALF of patients with IPF at the time of diagnosis suggests that increased expression of this factor is an early event in IPF. However, age of patients may be a confounding factor in the present study because patients with IPF were older. We could not study BALF NRG1α expression in the same patients who were included in the immunohistochemical study. In transgenic mice, activation of ErbB1 causes pulmonary fibrosis,3 while silencing of ErbB3 signalling in the lung protects mice against bleomycin-induced lung fibrosis.31 While the present studies support a potential role of NRG1α signalling in IPF, the mechanisms underlying differentiation, bronchiolarisation, and fibrosis are likely to be complex; signalling by the extracellular matrix may be important and other signalling molecules may be implicated. In patients with diffuse panbronchiolitis, polymorphisms of the MUC5B promoter are associated with increased promoter activity, increased MUC5B expression, and disease severity, suggesting that the differentiation of mucus cells towards a MUC5B+/MUC5AC- phenotype in IPF may be influenced genetically.32
Whether the acquisition of a mucus cell phenotype in the lungs of patients with IPF is an active differentiation process was not addressed in this study. Deletion of Munc13-2, a gene encoding an exocytic priming protein, causes the accumulation of MUC5B in the cytoplasm of Clara cells in mice;33 alterations of exocytic pathways may contribute to the acquisition of a mucus cell phenotype in the bronchiolised regions of IPF lungs. Cytoplasmic expression of HES1 was a constant feature of mucus cells, suggesting a role for the Notch pathway in mucus cell differentiation in the adult respiratory tract, although HES1 expression is also regulated by Notch-independent mechanisms.34
KLF-4, which regulates mucus cell differentiation in the gut, is highly expressed in the lungs of patients with rapidly progressive IPF.25 However, KLF-4 was not detected in the present study, suggesting that the mechanisms driving epithelial metaplasia in IPF may be specific to the respiratory system. Conversely, lack of KLF-4 detection in lungs with IPF may have been related to insufficient sensitivity of the antibody, post-translational regulation or selection bias.
Many features of the lesions seen in the peripheral lung in IPF were similar to those characteristic of proximal airway submucosal glands that express MUC5B rather than MUC5AC.18 NRG1α was found to be expressed in the cytoplasm of serous cells in submucosal glands, in proximity to p63-positive basal cells. P63-positive basal cells function as airway epithelial progenitor cells in humans.35 We speculate that NRG1α expression seen in lungs of patients with IPF may influence abnormal epithelial differentiation in cells derived from basal or other progenitor cells, resulting in mucus metaplasia with cellular features more typical of submucosal glands mucus cells.
In conclusion, our findings indicate that mucus cell differentiation in the distal airway epithelium of patients with IPF is distinct from that typical of the SPDEF-associated pathways seen in COPD, asthma or cystic fibrosis. The finding that NRG1α is increased in BALF in patients with IPF, and its role in regulation of mucus cell differentiation,14 support its potential paracrine effect influencing bronchiolisation and MUC5B expression.
Acknowledgments
The authors thank Dr Kathryn Wikenheiser-Grokamp for helpful suggestions, David Loudy, Gail Macke and Paula Blair for excellent technical assistance, and Ann Maher for secretarial help.
References
Supplementary materials
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Footnotes
See Editorial, p 647
Linked articles 161307.
Funding This work was supported by a travel grant from the Société de Pneumologie de Langue Française (LP), the National Institutes of Health HL095580 and HL090156, the Cystic Fibrosis Foundation RDP Center and the European Commission (FP7 grant agreement #202224, European IPF Network). Other Funders: NIH.
Competing interests None.
Ethics approval This study was conducted with the approval of the Justus-Liebig University of Giessen.
Provenance and peer review Not commissioned; externally peer reviewed.