Article Text

Abstract
Pulmonary disease is the most important cause of morbidity and mortality in cystic fibrosis (CF). Most patients with CF die from respiratory failure with extensive airway destruction. Airway remodelling, defined as structural airway wall changes, begins early in life in CF but the sequence of remodelling events in the disease process is poorly understood. Airway remodelling in CF has traditionally been thought to be solely the consequence of repeated cycles of inflammation and infection. However, new evidence obtained from developmental, physiological and histopathological studies suggests that there might instead be multiple mechanisms leading to airway remodelling in CF including (1) changes related to infection and inflammation; (2) changes specific to CF as a result of CF transmembrane conductance regulator (CFTR) dysfunction in the airway wall, independent of infection and inflammation; and (3) protective responses to (1) and/or (2). Recent advances in bronchoscopic techniques have allowed airway mucosal (endobronchial) biopsies to be taken in children and even infants. Endobronchial biopsy studies may provide insight into the role and relative contribution of the different mechanisms of airway remodelling in CF, with the main limitation that they assess only changes in proximal large airways and not in peripheral small airways from where CF disease is thought to originate. Findings from biopsy studies could encourage the development of novel therapeutic strategies targeting structural changes in addition to infection and inflammation.
- Cystic fibrosis
- paediatric lung disease
- airway remodelling
- endobronchial biopsy
- asthma
- bronchoscopy
- clinical epidemiology
- imaging/CT MRI etc
- paediatric asthma
Statistics from Altmetric.com
- Cystic fibrosis
- paediatric lung disease
- airway remodelling
- endobronchial biopsy
- asthma
- bronchoscopy
- clinical epidemiology
- imaging/CT MRI etc
- paediatric asthma
Introduction
Pulmonary disease is the most important cause of morbidity and mortality in cystic fibrosis (CF). It is characterised by extensive structural airway changes including bronchiectasis, bronchiectatic pus-filled cysts, mucoid impaction, atelectasis, fibrosis and vascular changes. These structural changes in the airway wall are referred to as remodelling which, in this context, has been defined as an alteration in size, mass or number of tissue structural components inappropriate to the maintenance of normal function.1 Airway remodelling may already be present in children with CF and, in more than 90% of patients with CF, ultimately leads to death from respiratory failure.2–4
To date, most work on the pathology of the airway wall in CF has involved the study of lungs taken at autopsy or removed prior to transplantation where there is advanced end-stage disease. Thus, little is known about disease pathogenesis in the airway during the early stages of CF. In this paper we review new evidence obtained from developmental, physiological and histopathological studies which provides information concerning the extent and development of airway remodelling in the early stages of CF and consider the implication of early structural changes in the CF lung in relation to the potential development of novel therapeutic strategies. We largely discuss human studies; relevant mouse models have recently been reviewed.5 6
Early structural changes to the CF airways
Autopsy studies performed more than two decades ago showed changes including squamous metaplasia, submucosal mucous gland enlargement and bronchiectasis in infants and young children with CF.2 3 However, the overall health of today's children with CF is much better and pulmonary health and survival of patients in successive birth cohorts have continuously improved.7 The histopathological changes observed in autopsy studies 20 or 30 years ago may not therefore reflect what is actually happening in the lungs of the children with CF we care for now.
Despite improved pulmonary health, airway disease in CF is present early, even in asymptomatic infants. Bronchoalveolar lavage (BAL) shows the presence of both infection and inflammation in infants with CF as young as a few weeks of age.8 CT scans also demonstrate the presence of remodelling in infants with CF.9–11 These studies indicate that remodelling occurs in the first months of life and includes thickened airway walls, narrowed airway lumens, air trapping and bronchiectasis. Lung function has also been shown to be diminished in infants with CF.12 13 One study reported that lung function (measured by the raised volume rapid thoracoabdominal compression technique) was preserved during the first 6 months of life, and proposed that this period may represent a ‘window of opportunity’ for therapeutic interventions.13 However, the number of subjects studied at this age was small.
Thus, structural changes to the airway wall in CF appear early in life. However, the sequence of remodelling events and their relationship to infection and inflammation remains unclear. Furthermore, the degree of reversibility, if any, of these early changes has not been determined.
Sequence of remodelling events in CF
Lessons learned from asthma
Airway remodelling in CF is traditionally thought to be solely the consequence of repeated cycles of inflammation and infection (figure 1). In asthma, a similar paradigm (ie, that chronic inflammation and repeated cycles of repair lead eventually to structural alteration of the airway wall) has recently been challenged.

(A) Traditional view of the sequence of events leading to airway remodelling in cystic fibrosis and (B) the model proposed by the authors. CFTR, cystic fibrosis transmembrane conductance regulator; RBM, reticular basement membrane.
Studies of endobronchial biopsies in asthma have shown that thickening of the reticular basement membrane (RBM) is already present in young wheezy children and that it is of the same magnitude as that seen in adults.14 Moreover, they have demonstrated that RBM thickening in children is not related to the duration of asthma, to treatment or to any inflammatory marker investigated. Furthermore, they have shown that, while there is no evidence of either eosinophilic inflammation or airway remodelling in infants with wheeze and reversible airflow obstruction at a median age of 1 year, both are present in preschool children with wheeze at a median age of 3 years.15 16 These findings suggest that inflammation and remodelling in asthma develop early in life and in parallel, not sequentially. This is supported by recent evidence from a novel neonatal mouse model of allergic airways disease which has shown that bronchial responsiveness, eosinophilic inflammation and remodelling occur in parallel during sequential airborne house dust mite sensitisation.17
Endobronchial biopsy as a tool to study airway remodelling early in life in CF
There is a paucity of data that shed light on the sequence of airway remodelling events in CF. In contrast to asthma where endobronchial biopsy has allowed detailed investigation of both airway remodelling and inflammation in the early stages, few groups have yet included endobronchial biopsy in their investigations of remodelling in CF (table 1).
Studies that have used endobronchial biopsy to investigate airway remodelling in cystic fibrosis (CF)
We have demonstrated the safety of endobronchial biopsy in infants and small children, including those with CF.26 The additional biopsy procedure takes only a few minutes, which we consider represents an acceptable increase in the duration of fibreoptic bronchoscopy for the purpose of research.27 In addition, we have reported that biopsy size and quality are adequate for the study of airway remodelling in children and infants with CF.28
There are, however, clearly limitations to this approach as the sample is only from (1) relatively proximal large airways rather than the distal small airways from where the earliest abnormal physiological signal originates29; (2) the airway mucosa of bronchial bifurcations (carinae) rather than the full thickness of the airway wall; and (3) an extremely small portion of the airway tree. The last-mentioned limitation is important because of intrasubject and interbiopsy variability and can be mitigated in part by sampling at least two sites.28
Provided studies are performed according to ethical guidelines and by experienced staff as part of focused hypothesis-driven research and processed adequately, biopsy studies may yield valuable new information about the pathogenesis of airway remodelling in CF children with early-stage lung disease (figure 2).24 25 30 We encourage consideration of endobronchial biopsy in children with CF, but only at the time of clinically-indicated bronchoscopy and subject to ethics, appropriate informed consent and the caveats listed above.31
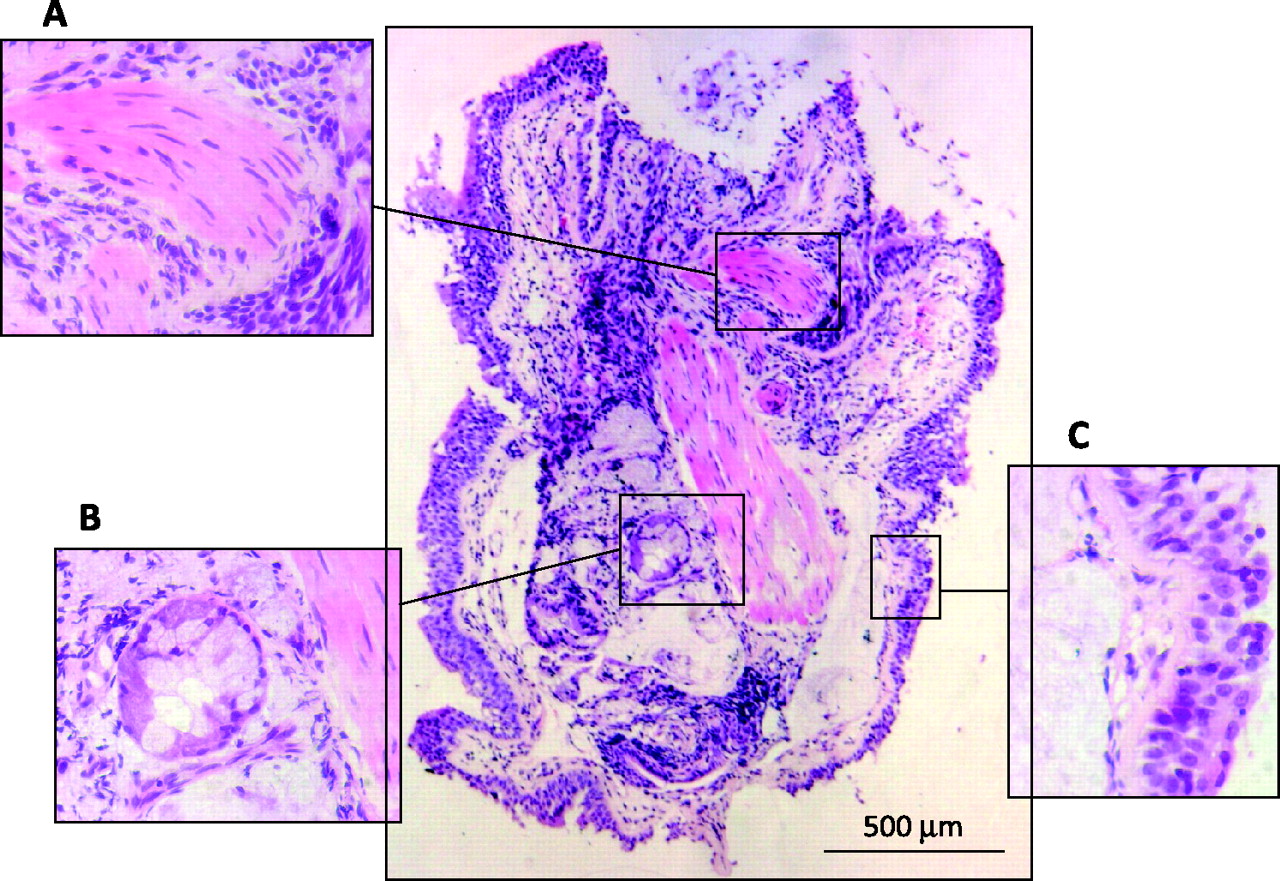
Endobronchial biopsy as a tool to study airway remodelling in cystic fibrosis (CF). Low power view of an endobronchial biopsy section obtained from a 4-year-old child with CF showing (A) smooth muscle; (B) glands; (C) epithelium. Stain: H&E.
Relationship of CF airway remodelling to inflammation and infection
As mentioned earlier, airway remodelling in CF is traditionally thought to be solely the consequence of repeated cycles of inflammation and infection. On the basis of current work we hypothesise that, in fact, there may be a number of mechanisms including (1) changes related to infection and inflammation; (2) changes specific to CF as a result of CF transmembrane conductance regulator (CFTR) dysfunction in the airway wall, independent of infection and inflammation; and (3) the result of protective responses to (1) and/or (2) (figure 1). If the hypothesis is correct, it is of more than academic importance. If remodelling in CF, eventually contributing to death, is only secondary to inflammation and/or infection, then early treatment of infection and inflammation should be an adequate therapeutic strategy alone. If there is a specific CFTR-related component, then different therapeutic strategies may be required. The following sections summarise available data on the relationships of CF airway remodelling to inflammation and infection.
Developmental data
The lungs of newborn infants with CF are thought to be essentially normal.32 However, dilatation of tracheal submucosal gland ducts, airway epithelial cells devoid of microvilli and increased levels of proinflammatory proteins have been described in CF fetuses, suggesting that subtle changes to the airways may occur prenatally.33–35 Because the fetus grows in a sterile environment, these changes are likely to be independent of infection and directly linked to CFTR dysfunction. However, if lung function is really normal very early in the life of newborn screened infants with CF (as has been suggested), then it is unlikely that antenatal remodelling is important.13
Physiological data
Physiological data (ie, respiratory function measures) have shown that the extent of airway obstruction in newly diagnosed infants with CF is unaffected by a history of lower respiratory illness, the presence of respiratory signs and symptoms, or the presence of pulmonary infection and inflammation in BAL fluid samples.12 13 36 Moreover, despite treatment of infection and inflammation, there is no ‘catch-up’ in airway function during infancy and early childhood or into the school years.12 36 37 Even in children with CF with no apparent respiratory complications, there appears to be a CF-specific airway obstruction suggestive of a CF-specific airway wall defect related to CFTR dysfunction and independent of infection and inflammation.12 36 37
Histopathological data
The interpretation of several previous studies of lung tissues has been hampered by two factors. First, tissue has come either from infants who died from meconium ileus (no or minimal lung disease) or from lungs at the time of transplantation or death where there is advanced end-stage disease. Second, the few studies that have used endobronchial biopsy to study early stages of the disease have been cross-sectional with no longitudinal data on airway remodelling (table 1). Thus, associations between inflammatory and structural changes have been described, but there are no data on causal relationships. We have used control groups of children with other chronic suppurative lung diseases (non-CF bronchiectasis such as primary ciliary dyskinesia) to try to tease out CF-specific changes from those that are secondary to inflammation.24 25 38
Epithelium and reticular basement membrane (RBM)
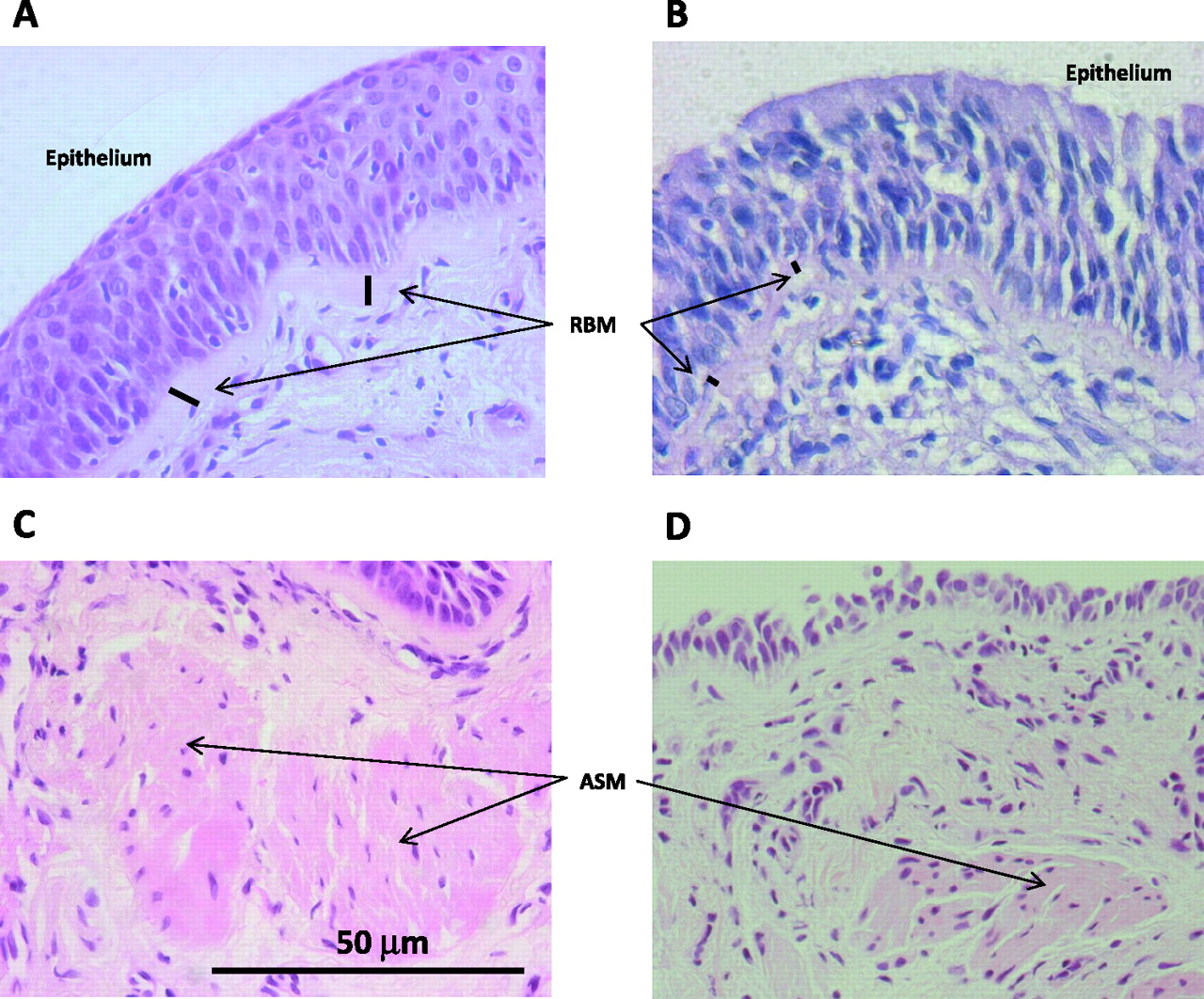
The thickness of the epithelial RBM has been studied at various stages of the disease. Studies in adults have yielded conflicting results; while some authors have reported RBM thickening,19 39 others have found that the RBM has been significantly thinner than normal.23 Repair processes (leading to RBM thickening) and neutrophil-mediated proteolytic degradation (leading to RBM thinning) have been proposed to explain these apparently opposing findings. It is also possible that reduced fibril synthesis late in the disease course may be important, but this is currently wholly speculative. In our studies we reported an increased RBM thickness in children with CF unrelated to the raised levels of inflammatory cells and proteins observed in BAL fluid but significantly related to BAL concentrations of transforming growth factor β1 (TGF-β1) (figure 3).24 These data indicate that this aspect of remodelling is independent of inflammation.
{kind=link}
{kind=link}
{kind=link}
Histopathological changes in the cystic fibrosis (CF) airways. High power view of representative endobronchial biopsy sections obtained from a subject with CF (A, C) and a control subject (B, D). Hyperplasia and squamous metaplasia of the epithelium, reticular basement membrane (RBM) thickening and increase in airway smooth muscle (ASM) content are seen in the CF biopsies. Stain: H&E.
The airway epithelium in adults with advanced CF disease exhibits hyperplasia and squamous metaplasia, loss of ciliated epithelial cells and ultrastructural abnormalities such as disorganisation of tight junctions and compound cilia (figure 3).2 18 32 40 41 However, it is not known whether these changes are CF-specific or simply secondary to inflammation and infection. Interestingly, prenatal loss of microvilli, atrophy and metaplasia of the tracheal epithelium with an abnormal Golgi apparatus and aberrant mitochondria have also been reported in autopsy tissue from fetuses with CF, suggesting that epithelial remodelling occurs prenatally and in the absence of infection.33 34 In support of this, a humanised nude mouse xenograft model shows delayed and abnormal epithelial regeneration in CF in the absence of bacterial infection.42 In addition, we have reported alterations in the respiratory and olfactory epithelium of CF mice, apparently unassociated with inflammation or bacterial infection.43
Extracellular matrix
There is evidence of increased airway tissue breakdown in CF. The presence of chondroitin sulfate and desmosine in sputum, raised levels of elastin and glycosaminoglycans in BAL fluid, raised levels of urinary desmosines and collagen metabolites, fragmented and exfoliated elastin in autopsy lung tissue and ultrastructural evidence of lysis of elastic and collagen fibres in endobronchial biopsies has been reported19 44 45; these probably represent degradation of important structural components of the extracellular matrix such as elastin, collagen and glycosaminoglycans. This breakdown of the airway tissue is likely to be functionally relevant, leading to a loss of lung function and favouring the development of bronchiectasis.24 44 Degradation of the bronchial tissue matrix is associated with an early protease/antiprotease imbalance in CF linked to inflammation and infection.24 44
Widespread fibrotic changes causing narrowing of the small airways have been described, especially in end-stage CF.3 4 19 However, little is known about the regulation of repair processes in response to proteolytic injury, the pathogenesis of fibrotic changes and the role of growth factors and their complex regulation in CF.46
Glands, mucins and goblet cells
Although dilation of tracheal submucosal gland ducts has been described in CF fetuses,34 early autopsy lung specimens from infants with CF have shown that mucus-secreting submucosal glands are in general normal in both size and number, regardless of the presence of airway infection.32 However, there is an enlargement of submucosal glands in older patients.2 3 Thus, although patients with CF seem to be born with, at most, only minor mucous cell abnormalities, major changes occur by the time of death. Recent bronchoscopic data have shown an increase in submucosal gland volume with proportionate increases in both serous and mucous cell components in adults with CF.23 As submucosal glands have been found more distally (ie, in smaller airways) in CF lungs at autopsy than in controls, it seems likely that, at least in part, gland enlargement in CF results from growth (hyperplasia) of new submucosal gland cells.23 The exact mechanisms behind this gland enlargement in CF are unknown, but neutrophil-directed epithelial cell activation leading to epithelial mesenchymal transition, epithelial tubulogenesis and new gland formation has been proposed.23
Gel-forming mucins such as MUC2, MUC5AC and MUC5B are secreted by airway mucous cells. They are subject to regulation by inflammatory stimuli and contribute to airway plugging in CF.47 However, studies on mucin expression and secretion in CF have yielded conflicting results. While some authors have reported increased expression or secretion of mucins in CF,47 others have found the opposite.48 An increase in the volume of stored mucin in the epithelium, resulting in increased goblet cell size rather than number, has been shown in airway tissue obtained at transplant and in endobronchial biopsies from subjects with CF.21 23 47 Similarly, an increase in the volume of stored mucin in submucosal glands has been demonstrated.21 23 However, it remains controversial as to whether the increase in mucin stores represents increased mucin production and secretion that contributes to luminal plugging,47 or whether mucin secretion is slowed in CF resulting in its retention within goblet cells and glands and a lack of a protective mucous layer which would predispose the CF airway to subsequent infection.48
Smooth muscle, cartilage and vessels
There is an increase in airway smooth muscle (ASM) content in lungs (autopsy, explants or lobectomy) from adults with severe end-stage disease, as well as in biopsies obtained from CF adults with mild to moderate airway obstruction.22 40 Using endobronchial biopsies we have recently reported increases in ASM cells (both number and size) in children with CF (figure 3).25 Interestingly, these changes were also present in children with other inflammatory lung diseases such as asthma and non-CF bronchiectasis, suggesting common (not CF-specific) pathophysiological changes probably related to airway inflammation and/or infection. The cellular mechanisms leading to these structural changes are incompletely investigated, but it has been proposed that an increase in ASM could contribute to airway hyper-responsiveness.40 49
Loss of cartilage resulting in tracheomalacia and bronchomalacia and related to the severity of bronchiectasis is found in end-stage CF disease of the airways.40 50 51 It is associated with chronic inflammation and infection and is preceded by destruction and fibrotic replacement of the cartilage. There is also increased bronchial blood flow and vascular remodelling in CF. The causes and mechanism(s) are unknown, but endothelial dysfunction—which is frequent in end-stage CF and involves the NF-κB pathway implicated in inflammation—is thought to precede the vascular changes.52
Bronchial mucosal inflammation
The most characteristic feature of pulmonary inflammation in CF is the predominance of large numbers of neutrophils in the airway lumen.2–4 8 32 This excess of neutrophils is considered part of a ‘vicious cycle’ of increased inflammation: neutrophils release an array of mediators, oxidants and proteases including neutrophil elastase which play a major role in the pathophysiology of chronic inflammation and tissue damage. The neutrophils appear to be ineffective at clearing the airway of pathogens, perhaps due in part to the cleavage of CXCR1 motifs from their surface by elastase.53 BAL studies have shown that neutrophilic inflammation occurs very early in the disease.8 Whether inflammation is a primary event linked to CFTR dysfunction or only follows infection remains unclear.8
The investigation of airway lumen events has not been paralleled by studies focused on the airway wall. Indeed, very little is known about the recruitment and accumulation of inflammatory cells in the tissues of patients with CF, and particularly that which occurs in the bronchial mucosa, one site where airway remodelling takes place. Early pathological studies have reported inflammation in the bronchial wall in children as young as a few months of age.2 3 32 Quantitative data of inflammatory cells in the airway wall at the time of transplantation have shown an accumulation of lymphocytes, which consisted mainly of T lymphocytes, especially distally where intense tissue damage is observed.4 39 Lymphoid aggregates formed by B cells are also observed along the length of CF airways. In contrast, neutrophils appear to be localised mainly to the surface epithelium, suggesting a preferential migration of these cells towards the airway lumen.39 Only one study has assessed the inflammatory infiltrate occurring during earlier stages of CF lung disease; using endobronchial biopsies, Wojnarowski et al described a predominantly lymphocytic infiltrate in the airway subepithelium in children with CF with stable disease in contrast to the neutrophil-dominated infiltrate in children with CF with an acute pulmonary exacerbation.20 Thus, lymphocytes may play a role in the pathophysiology of CF lung disease, which we are currently investigating further.54
Is there a CFTR-specific airway defect?
Currently this is speculative. Lines of evidence that are supportive of this concept are the findings of (1) subtle prenatal changes in CF fetuses33–35; (2) airflow obstruction in children with CF who at diagnosis have no discernable respiratory complications, which persists into the school years even in the absence of respiratory complications and despite specialist treatment12 13 36 37; and (3) histopathological changes including delayed and abnormal epithelial regeneration, epithelial thickening, increased RBM thickness and mucus cell number that appeared to be independent of infection and inflammation.24 42 43
Functional consequences and reversibility of airway remodelling in CF
While parenchymal destruction is clearly detrimental to lung function,24 44 other changes of remodelling might in fact be protective. RBM thickening, for instance, might protect the airway by limiting the access of elastases and other proteolytic enzymes released from necrotic neutrophils in particular to the subepithelial compartment.55 It might also be protective by preventing or ameliorating airway narrowing and thus minimising air trapping and hyperinflation. As reviewed in an earlier section, both RBM thickening and thinning have been reported in CF. It has been suggested that repair processes and neutrophil-mediated proteolysis may well have opposing effects on RBM thickness; thus, RBM thickening may be protective in the early stages of the disease but, subsequently, the initially protective response becomes overwhelmed and the RBM thins owing to an increased burden of elastase.19
There are no data on reversibility of airway remodelling in CF. In asthma, studies of endobronchial biopsies have demonstrated reversal upon pharmacological intervention of some components of airway remodelling such as reduction of RBM thickening or of increased airway vascularity.56 57
Conclusions
Airway remodelling in CF appears early in life and the window of opportunity for prevention of the onset of progressive airway damage that characterises CF lung disease may be short. Current strategies focus on the prevention and treatment of infection and inflammation. However, we suggest that, although important, this by itself may be insufficient. There is some evidence of CFTR-specific airway wall dysfunction and also of structural changes that may be protective; modulation of both of these may offer new therapeutic avenues.
As cross-sectional data can at best only demonstrate hypothesis-generating associations, there is a major need in the future for longitudinal studies; inclusion of endobronchial biopsy affords this possibility. We have confirmed that endobronchial biopsy is safe and feasible even in very young children with CF and provides us with the means to understand better the early mechanisms of airway remodelling. Although there is currently no clinical indication for performing endobronchial biopsy in children with CF, we would encourage units which routinely perform annual surveillance bronchoscopy in CF to consider performing endobronchial biopsy for research as part of this procedure so that these hypotheses can be tested.
References
Footnotes
Funding NR is the recipient of a European Respiratory Society Fellowship (Nr. 64).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.