Article Text

Abstract
Background: Despite intense research efforts, the aetiology and pathogenesis of idiopathic pulmonary fibrosis remain poorly understood. Gelsolin, an actin-binding protein that modulates cytoskeletal dynamics, was recently highlighted as a likely disease modifier through comparative expression profiling and target prioritisation.
Methods: To decipher the possible role of gelsolin in pulmonary inflammation and fibrosis, immunocytochemistry on tissue microarrays of human patient samples was performed followed by computerised image analysis. The results were validated in the bleomycin-induced animal model of pulmonary inflammation and fibrosis using genetically-modified mice lacking gelsolin expression. Moreover, to gain mechanistic insights into the mode of gelsolin activity, a series of biochemical analyses was performed ex vivo in mouse embryonic fibroblasts.
Results: Increased gelsolin expression was detected in lung samples of patients with idiopathic interstitial pneumonia as well as in modelled pulmonary inflammation and fibrosis. Genetic ablation of gelsolin protected mice from the development of modelled pulmonary inflammation and fibrosis attributed to attenuated epithelial apoptosis.
Conclusions: Gelsolin expression is necessary for the development of modelled pulmonary inflammation and fibrosis, while the caspase-3-mediated gelsolin fragmentation was shown to be an apoptotic effector mechanism in disease pathogenesis and a marker of lung injury.
Statistics from Altmetric.com
Epithelial damage or apoptosis is a common feature of pulmonary pathophysiology, including infection, acute lung injury and interstitial diseases.1 With regard to idiopathic pulmonary fibrosis (IPF), the most common idiopathic interstitial fibrotic pulmonary disease,2 recent studies strongly suggest a role for epithelial apoptosis as a key profibrotic event.3 Apoptosis of alveolar epithelial cells is found in the lungs of patients with IPF as well as in animal models of the disease, correlating with the increased expression of “death inducing” tumour necrosis factor/tumour necrosis factor receptor family members and various apoptotic markers.1 3–9 Furthermore, induction of epithelial apoptosis is sufficient to initiate a fibrotic response in animal models,10 whereas genetic or pharmacological blocking of apoptotic signals can prevent a bleomycin (BLM)-induced fibrotic response.11 These observations have contributed significantly to the prevailing hypothesis that the mechanisms driving IPF reflect abnormal deregulated wound healing in response to multiple sites of ongoing alveolar epithelial injury.3
Remodelling the actin cytoskeleton in response to stress, such as apoptotic stimuli, is a fundamental process in many eukaryotic cells that regulates cell morphology, polarity, motility and division.12 The assembly and disassembly of actin filaments, as well as their organisation into functional high order networks, are regulated by several actin-binding proteins.13 Among them, gelsolin (GSN) is an actin filament capping/severing protein that has been implicated, among others, in the transduction of signals into dynamic rearrangements of the cytoskeletal architecture.14 Moreover, caspase-3 mediated GSN fragmentation15 has been shown to contribute to actin cytoskeletal collapse and apoptosis through the amino terminal GSN cleavage product.16
Gsn expression has been found deregulated in various types of cancer and oncogenic transformations,14 and was selected as a likely disease modifier in IPF through comparative expression profiling and target prioritisation.6 To explore the possible role of GSN in the development of pulmonary inflammation and fibrosis, we first examined its expression in tissue microarrays of lung samples from patients with idiopathic interstitial pneumonia (IIP). Increased Gsn expression was noted in the pulmonary epithelium as well as in BLM-induced pulmonary inflammation and fibrosis, where it was shown to be necessary for disease development. Disease protection was attributed to defective neutrophil infiltration and decreased epithelial apoptosis, where a series of bone marrow transfers indicated attenuated epithelial apoptosis in the absence of GSN as the primary cause of disease protection.
METHODS
Patients
Lung tissue samples from 65 untreated patients were isolated by open lung biopsy or video-assisted thoracoscopic surgery for diagnostic purposes, according to the ATS/ERS criteria.17 Following protocol approval by the local ethics committee (#1669), all patients signed an informed consent form where they agreed to the anonymous usage of their lung samples for research purposes. 70% of the patients had also been included in a previous study.6 Lung samples were used for the construction of tissue microarrays.18 The construction of tissue microarrays, immunostaining, computerised image analysis and statistical analysis has been described previously.6
Animals
The generation of gsn−/− mice by homologous recombination in embryonic stem (ES) cells has been described previously.19 All mice strains were bred and maintained in the 129×C57/Bl6 background for over 10 generations at the animal facilities of the Alexander Fleming Biomedical Sciences Research Institute under specific pathogen-free conditions in compliance with the Declaration of Helsinki principles. The isolation of mouse embryonic fibroblasts from gsn−/− and wild-type (WT) littermate mice is described in the online supplement.
BLM-induced pulmonary inflammation and fibrosis
Pulmonary fibrosis was induced by a single tail vein injection of bleomycin hydrogen chloride (100 mg/kg body weight; 1/3 LD50; Nippon Kayaku, Tokyo) to 6–8-week-old mice as previously reported in detail.4 Lung histology and immunocytochemistry, broncheoalveolar lavage (BAL) fluid collection and analyses, inflammatory cell count and collagen measurements as well as bone marrow transfers were performed as previously described.4
LPS-induced acute lung injury
10 mg of bacterial lipopolysaccharide (LPS) from Pseudomonas aeruginosa (Sigma, St Louis, Missouri, USA) was dissolved in normal saline at a final volume of 3 ml. The solution was administered by a custom-made nebuliser flowing at 4 l/min oxygen for 20–25 min into an airtight chamber containing 5–7 mice. All analyses were performed 24 h later.
Assays
Chemokine (C-X-C motif) ligand 1 (Cxel1; KC), ELISA and terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) assays were performed as previously described.4 Myeloperoxidase (MPO), caspace-3 activity, plasmid degradation, α-GSN western blots, immunocytochemistry assays and real-time RT-PCR analysis are described in the online supplement.
RESULTS
Increased GSN expression in human fibrotic lungs
To explore a possible role for GSN in the pathogenesis of pulmonary inflammation and fibrosis, its expression was examined in lung samples of patients diagnosed with IIP according to the ATS/ERS criteria (table 1).17
To expedite and standardise experimental procedures, two tissue microarrays18 were created using the following patient lung samples: 25 IPF/usual interstitial pneumonia (IPF/UIP), 12 fibrotic non-specific interstitial pneumonia (fNSIP), 8 cellular non-specific interstitial pneumonia (cNSIP) and 20 cryptogenic organising pneumonia/organising pneumonia (COP/OP); 2–3 tissue cores per patient paraffin sample were embedded in recipient paraffin blocks together with 20 control samples derived from normal areas of lungs removed for benign lesions. Tissue microarray sections were immunostained for GSN and signal intensities were analysed qualitatively by blind microscopy (fig 1A and fig E1 in online supplement) and quantitatively/statistically by computerised image analysis (fig 1B).
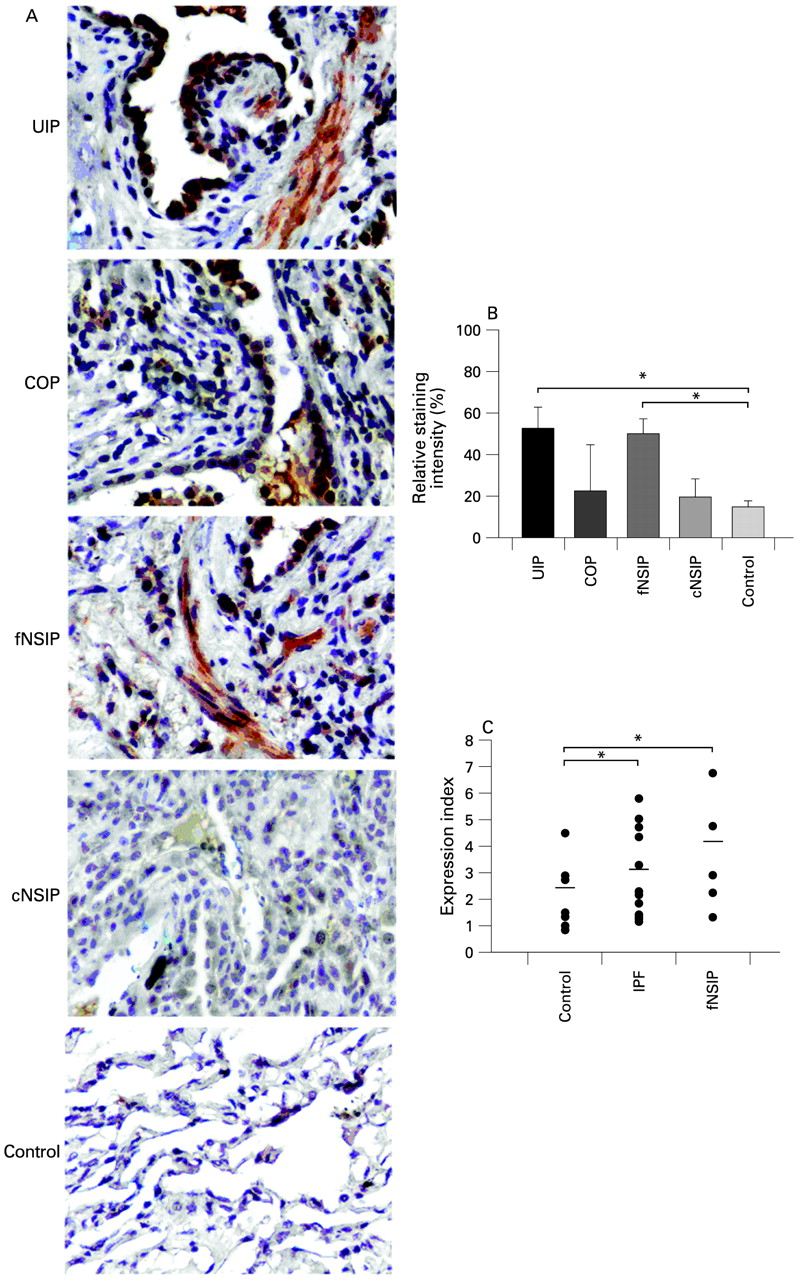
Increased gelsolin (GSN) expression within the fibrotic lung. (A) Representative immunocytochemistry with an α-GSN antibody on tissue microarrays containing 25 IPF/UIP, 20 COP, 20 NSIP and 20 normal lung samples as described in table 1. (B) Computerised image analysis of immunostained sections. Data are presented as bar charts of mean (SD) values depicted by error bars. *p<0.05 between the indicated groups (unpaired t test). (C) Real-time PCR on total RNA isolated from frozen lung samples of (different groups of) patients with interstitial idiopathic pneumonia also revealed an increase in GSN expression. Values were normalised to the corresponding values of the reference gene b2m and presented as expression index. Actual data are presented with lines indicating mean values. *p<0.05 between the indicated groups (t test). COP, cryptogenic organising pneumonia; IPF, idiopathic pulmonary fibrosis; cNSIP, cellular non-specific interstitial pneumonia; fNSIP, fibrotic non-specific interstitial pneumonia; UIP, usual interstitial pneumonia.
Significant expression of GSN was observed in IPF/UIP and fNSIP samples and was almost missing from normal lung control samples (fig 1A and B). Similar results were also obtained by real-time RT-PCR analysis (on a different set of IIP samples processed independently (fig 1C). GSN exhibited strong positive staining within areas of the epithelium (bronchial and alveolar) and to a lesser extent in adjacent fibroblastic foci within the IPF lung. GSN was localised in areas of active fibrosis and the surrounding alveolar epithelium in fNSIP, and was also present within the alveolar epithelium overlying areas of active fibrosis called Masson bodies. It showed weak positive staining in the fibrotic interstitium of the COP lung compared with IPF and fNSIP samples (fig 1A and fig E1 in online supplement). Quantification and statistical analysis of signal intensities of all 85 samples revealed an overall increase in GSN expression in IPF/UIP and fNSIP samples compared with cNSIP, COP/OP and normal samples (fig 1B), suggesting a role for GSN in disease pathogenesis.
GSN expression is necessary for the development of modelled pulmonary inflammation and fibrosis
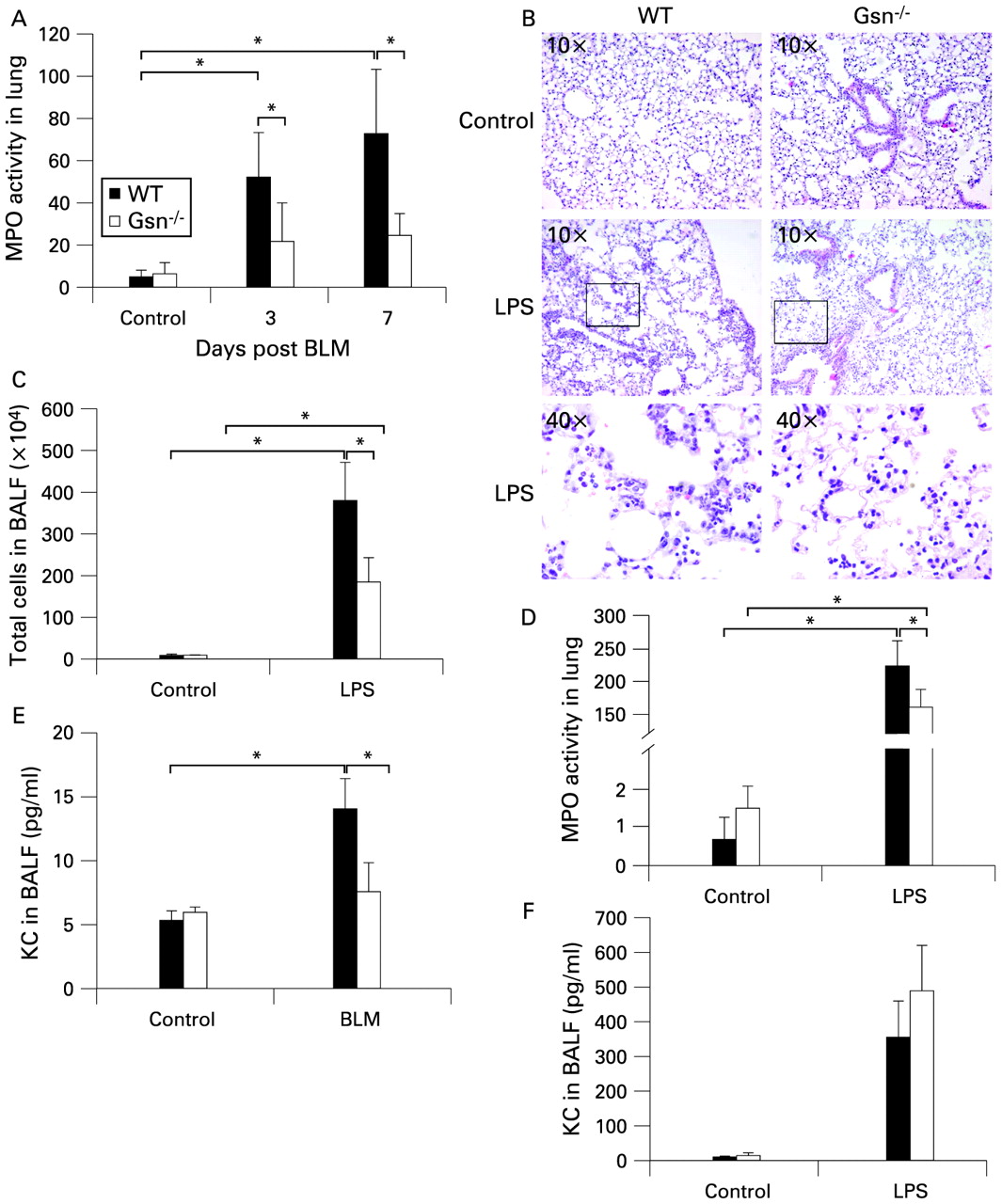
To confirm the possible role of GSN in the development of pulmonary inflammation and fibrosis, we used the animal model of BLM-induced pulmonary inflammation and fibrosis. As previously shown in detail elsewhere,4 BLM administration in WT mice resulted in progressive subpleural/peribronchial pulmonary inflammation which subsequently diffused into the parenchyma (fig 2A). Accordingly, inflammatory cells in BAL fluid increased gradually after BLM injection (fig 2B). Inflammation was followed by the development of mainly subpleural and peribronchial fibrotic patches characterised by alveolar septa thickening and focal dilation of respiratory bronchioles and alveolar ducts (fig 2A). Concomitantly, collagen production measured in lung protein extracts with a soluble collagen assay was increased (fig 2C). BLM administration resulted in a moderate increase in GSN levels in the lung interstitium, as measured by immunocytochemistry, mainly in epithelial (and endothelial) cells (fig 2D). Unlike the human samples, no staining was detected in fibroblasts, possibly reflecting (patho)physiological and technical differences (as mouse fibroblasts are hardly visible with H&E staining and hardly react to specific immunostaining) between mouse and human lungs. The increased epithelial GSN expression was confirmed by co-localisation with epithelial specific markers (T1A for type I and SPC for type II; fig E2 in online supplement). Increased GSN levels were also detected in the airspaces following BLM administration using an ELISA assay in the corresponding BAL fluid samples (fig 2E).

Gsn deficiency protects mice from bleomycin (BLM)-induced pulmonary fibrosis. Wild-type (WT) and Gsn−/− mice were injected intravenously with saline or BLM and killed at the indicated time points following injection. (A) Representative H&E stains (4×) of WT and Gsn−/− lung sections. Inflammatory infiltrates (open arrows) and fibrosis development (closed arrows) are evident in WT lungs in subpleural and peribronchial areas, whereas Gsn−/− mice show minimal signs of the disease. (B) Total inflammatory cell counts in bronchoalveolar lavage fluid (BALF). (C) Soluble collagen determination in lung extracts. (D) Immunodetection of gelsolin (GSN) in paraffin sections showing increased expression following BLM injection, mainly in epithelial cells (see also fig E2 in online supplement). (E) GSN levels were significantly increased in BALF samples from BLM-injected WT mice, determined by an ELISA assay. Data are presented as bar charts of mean (SD) values depicted by error bars (B and C) or as actual data with lines indicating mean values (E). *p<0.05 between the values of the indicated groups (t test, n = 3–5).
To determine the role of GSN in the development of pulmonary inflammation and fibrosis, we next administered BLM to genetically-modified mice lacking GSN.19 Gsn−/− mice were protected from development of the disease, exhibiting normal lung architecture, no inflammation and lack of collagen accumulation (fig 2A–C).
Impaired pulmonary neutrophil infiltration in Gsn−/− mice
As GSN was shown to be necessary for the development of pulmonary inflammation and fibrosis, its role in different cell compartments was further studied. To assess neutrophil infiltration in the lungs of Gsn−/− mice, myloperoxidase (MPO) levels in lung extracts were measured at early time points following BLM administration using the MPO assay. As shown in fig 3A, MPO levels in the lungs of WT mice were significantly increased at both 3 and 7 days after BLM administration, in sharp contrast to Gsn−/− mice. To verify the defective recruitment of Gsn−/− neutrophils into the lung upon injury, aerosolised LPS—a potent inducer of acute lung injury and neutrophil chemotaxis—was administered to WT and Gsn−/− mice. Histological examination of lung sections from WT mice 24 h after LPS administration revealed thickening of the alveolar septa and infiltration of inflammatory cells, consisting mainly of neutrophils (fig 3B). In contrast, Gsn−/− lungs had less evidence of acute lung injury with fewer infiltrating neutrophils. Both the absolute numbers of cells in the BAL fluid (>90% neutrophils) and the MPO levels were significantly reduced in the lungs of Gsn−/− mice (fig 3C and D).

Impaired neutrophil migration in the lungs of Gsn−/− mice. (A) Administration of bleomycin (BLM) resulted in increased neutrophil infiltration in the lungs of wild-type (WT) mice compared with Gsn−/− mice, as shown by myeloperoxidase (MPO) assays. (B) Representative H&E stain of lung sections of WT and Gsn−/− mice 24 h after administration of aerosolised lipopolysaccharide (LPS) showing increased sequestration of inflammatory cells (mainly neutrophils) in WT lungs, an effect markedly impaired in Gsn−/− lungs. (C, D) Reduced neutrophil infiltration in Gsn−/− lungs after exposure to LPS was also detected with cell counts in cytospins (C) and MPO assays (D). (E, F) Significantly reduced bronchoalveolar lavage fluid (BALF) levels of chemokine (C-X-C motif) ligand 1 (KC) were noted in Gsn−/− lungs 7 days after BLM injection (E), while no differences were found between the two mouse strains after LPS administration (F) as determined by an ELISA assay. Data are presented as bar charts of mean (SD) values depicted by error bars. *p<0.05 between the values of the indicated groups (t test, n = 3–6).
As the attenuated neutrophil recruitment could be directed from aberrant chemotactic signals, the levels of KC (CXCL1; the equivalent of human interleukin-8), a potent and essential neutrophil chemoattractant,20 were also examined. KC expression after BLM administration was found to be significantly reduced in Gsn−/− mice, both at the mRNA level (data not shown) and the protein level (fig 3E). No difference in KC levels was observed upon LPS administration (fig 3F).
Attenuated pulmonary epithelial apoptosis in Gsn−/− mice
As GSN has been implicated in caspase-3 mediated apoptotic mechanisms,15 21 the degree of apoptosis in the lungs of BLM-challenged mice was assessed. TUNEL assays, which detect primarily apoptotic cells,22 performed on lung sections 7 days after BLM injection, revealed alveolar epithelial cell apoptosis which was significantly lower in Gsn−/− mice (fig 4A and B). Reduced epithelial TUNEL+ cells in Gsn−/− mice were also observed when lung injury was induced with inhaled LPS (fig 4C and D), which has been known to induce epithelial apoptosis.23

Gsn deficiency results in reduced epithelial apoptosis upon lung injury. (A) Bleomycin (BLM) induced an increase in TUNEL+ cells in the lungs of wild-type (WT) mice, a response abrogated in Gsn−/− lungs. (B) Results expressed as the number of TUNEL+ cells per optical field in eight different areas per animal. (C) TUNEL assays performed on lung sections 24 h after administration of aerosolised lipopolysaccharide (LPS) revealed fewer apoptotic cells in the lungs of Gsn−/− mice compared with WT mice. (D) Results expressed as the number of TUNEL+ cells per optical field in eight different areas per animal. (E) WT and Gsn−/− lungs exhibited equal activation of caspase-3 after exposure to BLM, as judged by a corresponding activity assay. Values were expressed normalised to the corresponding saline controls. (F) Both BLM and LPS administration resulted in the caspase-3 mediated cleavage of full length GSN (FL-GSN) and the generation of the 41 kD COOH terminal (C-GSN) and the 39 kD NH2 terminal (N-GSN) fragments. Actin was used as a loading control. Data are presented as bar charts of mean (SD) values depicted by error bars (B and D) or as actual data with lines indicating mean values (E). *p<0.05 between the values of the indicated groups (t test, n = 3–7).
To investigate if the observed reduced apoptosis in Gsn−/− mice could be attributed to defective caspase-3 activation upon injury, lung extracts isolated 7 days after BLM administration were assayed for caspase-3 activity with an enzymatic assay (fig 4E). WT and Gsn−/− mice had equal levels of caspase-3 activity following administration of BLM, confirming the pro-apoptotic role of BLM and suggesting that the observed differences in apoptosis on GSN ablation lie downstream.
To determine if caspase-3 GSN fragmentation, a suggested apoptotic effector mechanism,15 is involved in BLM-induced lung injury and pulmonary inflammation and fibrosis, lung extracts isolated 7 days after BLM administration were immunoblotted for GSN. As shown in fig 4F, BLM administration provoked GSN fragmentation, an effect also observed with LPS administration.
Moreover, a series of biochemical analyses in WT and Gsn−/− littermate mouse embryonic fibroblasts (see online supplement and figs E3 and E4) confirmed that GSN ablation results in reduced apoptosis and is correlated with delayed disassembly of the actin cytoskeleton, as well as with reduced nucleolytic activity.
Gsn ablation in the stromal compartment is sufficient to inhibit disease development
Disease protection in Gsn−/− mice was correlated with both attenuated neutrophil infiltration and reduced epithelial apoptosis. To examine their relative contribution in disease development, reciprocal bone marrow transfer experiments were performed as previously described in detail.4 Properly reconstituted recipient mice (data not shown) bearing haematopoietic cells with the genetic background of the donor mouse and radioresistant/stromal cells with the genetic background of the host mouse were then injected with BLM to assess disease development. Mice that lacked Gsn expression in the haematopoietic compartment and therefore neutrophils (Gsn−/− to WT) developed both inflammation and fibrosis, as seen in the histological examination, inflammatory cell count and collagen levels (fig 5A–C). On the other hand, abolishing Gsn expression from the radioresistant stromal cells (WT to Gsn−/−) resulted in complete protection against disease (fig 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Gelsolin (GSN) ablation from the stromal compartment confers resistance to bleomycin (BLM)-induced pulmonary inflammation and fibrosis. Reciprocal bone marrow transfer experiments show that the absence of GSN from the radioresistant stromal cellular compartment protects mice from BLM-induced pulmonary inflammation and fibrosis. (A) Representative H&E stains (4×). (B) Total inflammatory cell counts in bronchoalveolar lavage fluid (BALF). (C) Soluble collagen determination in lung extracts. Data are presented as bar charts of mean (SD) values depicted by error bars. *p<0.05 between the values of the indicated groups (t test, n = 6–8).
DISCUSSION
In this study GSN expression was found to be increased in the fibrotic lungs of patients with IIP, suggesting a role in the pathogenesis of pulmonary inflammation and fibrosis. GSN expression was shown to be necessary for disease development, as Gsn−/− mice were protected from the modelled disease attributed to attenuated neutrophil infiltration, reduced chemotaxis and reduced epithelial apoptosis. BLM-induced apoptosis in vivo was shown to provoke caspase-3 mediated GSN fragmentation, a finding also confirmed ex vivo. A series of bone marrow transfers indicated GSN deficiency in stromal cells as the primary cause of the observed protection against disease.
IIP is a heterogeneous group of diseases consisting of seven distinct clinical and pathological entities.24 IPF is the most common IIP and has the same poor prognosis as fNSIP, both being unresponsive to corticosteroids. On the other hand, cNSIP and COP have a more favourable course and response to treatment.25 GSN was found to be overexpressed in patients with IPF and fNSIP compared with control subjects and those with cNSIP and COP (fig 1 and fig E1 in online supplement), suggesting a role for GSN in disease pathogenesis. Furthermore, the distinct GSN expression profiles in various types of IIPs may explain the differences in disease progression and response to treatment.
To determine the possible role of GSN in pulmonary inflammation and fibrosis, we have used the animal model of BLM, an experimental approximation of the human disease. Gsn−/− mice were found to be protected from disease development (fig 2); disease protection was correlated with both attenuated neutrophil infiltration (fig 3) and reduced epithelial apoptosis (fig 4).
Neutrophil infiltration represents the first line of defence of the immune system against infection or injury and, although such an acute inflammatory event cannot be assessed in patients with IPF, neutrophil infiltration in the course of BLM-induced pulmonary inflammation and fibrosis is well documented.26 GSN ablation resulted in attenuated neutrophil infiltration in the lung following BLM (or LPS) treatment (figs 2 and 3), in accordance with previous studies showing a delayed response to intraperitoneal thioglycollate.19 The blunted neutrophil response in the absence of GSN is not dependent on the levels of chemoattractive signals such as KC (the secretion of which was found to be decreased in the absence of GSN; fig 3), as Gsn−/− neutrophils failed to migrate efficiently towards the very potent neutrophil chemotactic peptide n-formyl-nle-leu-phe (nFMLP) both in vitro19 and in vivo when administered intratracheally and in parallel with BLM administration (data not shown). Likewise, LPS-induced neutrophil infiltration in Gsn−/− mice was reduced, despite similar levels of KC secretion (fig 3). Interventions that inhibit neutrophil migration and/or activation in BLM-induced pulmonary inflammation and fibrosis have resulted in amelioration of disease severity but have proved inadequate for complete protection.27 28 Likewise, GSN deficiency in haematopoietic cells leading to impaired neutrophil infiltration did not protect mice from BLM-induced disease development (fig 5). As GSN has been implicated in the transduction of signals into dynamic rearrangements of the cytoskeletal architecture,14 it seems very likely that, in the absence of the nucleating/severing activity of GSN, a less dynamic cytoskeleton in neutrophils would be unable to respond effectively to chemotactic cues.
Besides its important role in mediating actin cytoskeletal rearrangements, GSN is a substrate of caspase-3,15 whose inhibition has been shown to attenuate BLM-induced pneumonopathy.11 As shown in this study, BLM treatment induced caspase-3 activation both in vivo (fig 4) and ex vivo (fig E3 in online supplement), leading to poly-(ADP-ribose) polymerase (PARP)29 and GSN cleavage and epithelial apoptosis (fig 4). GSN genetic ablation attenuated epithelial apoptosis despite equal caspase-3 activation and PARP cleavage (fig 4). Moreover, GSN ablation resulted in complete disease protection only in radioresistant stromal cells (fig 5). Although the absence of GSN could have affected other pulmonary cell types differently with important implications for the development of pulmonary inflammation and fibrosis—such as endothelial cells30 and fibroblasts19 31—neither of these cells can be primarily affected by BLM because, unlike epithelial cells,32 they both possess BLM hydrolase which catabolises BLM. Disease protection resulting from GSN deficiency in stromal cells can therefore only be attributed to reduced epithelial apoptosis in the absence of GSN, suggesting an apoptotic effector role for GSN fragmentation in BLM-induced lung injury in vivo. GSN fragmentation was also observed with LPS challenge, suggesting that GSN fragmentation is a common caspase-3 apoptotic mechanism in lung injury.
Intracellular GSN and GSN-mediated responses were shown to be important in the pathogenesis of pulmonary inflammation and fibrosis. A structurally distinct soluble form of GSN is secreted in the plasma (pGSN) and has been shown to dissociate DNase I from extracellular actin and to bind and inactivate LPS.33 More importantly, pGSN binds and presents lysophosphatidic acid to cells,34 and activation of its receptor in lung fibroblasts was recently shown to be a prerequisite for the development of BLM-induced disease.35 Increased levels of GSN were detected in BAL fluid samples from mice after BLM treatment, although its origin remains unclear as it could have been released from damaged cells following injury, secreted from epithelial cells or extravasated from the blood.
In conclusion, increased GSN expression was detected in patients with IPF and fNSIP, possibly predicting response to treatment. Intracellular GSN was found to be necessary for efficient neutrophil infiltration, while its fragmentation was shown to be an apoptotic effector mechanism in epithelial apoptosis, a prerequisite for BLM-induced lung injury. Controlling epithelial cytoskeletal dynamics with actin-modifying drugs or targeting N-GSN could prove useful in controlling pulmonary inflammation and fibrosis.
Acknowledgments
The authors thank Peggy Andriopoulou for excellent technical assistance and Dr R M DuBois for critical reading of the manuscript.
REFERENCES
Footnotes
Competing interests: None.
Funding: European Commission Network of Excellence grant QLRT-CT-2001-01407 and a Hellenic Ministry for Development grant GSRT-PENED-136.
Ethics approval: Following protocol approval by the local ethics committee (#1669), all patients signed an informed consent form where they agreed to the anonymous usage of their lung samples for research purposes. All experimentation was approved by an internal Institutional Review Board, as well as by the Veterinary Service and Fishery Department of the local governmental prefecture.
See Editorial, p 461
▸ Additional methods data are published online only at http://thorax.bmj.com/content/vol64/issue6