Article Text

Abstract
Background There is microscopic spatial and temporal heterogeneity of pathological changes in idiopathic pulmonary fibrosis (IPF) lung tissue, which may relate to heterogeneity in pathophysiological mediators of disease and clinical progression. We assessed relationships between gene expression patterns, pathological features, and systemic biomarkers to identify biomarkers that reflect the aggregate disease burden in patients with IPF.
Methods Gene expression microarrays (N=40 IPF; 8 controls) and immunohistochemical analyses (N=22 IPF; 8 controls) of lung biopsies. Clinical characterisation and blood biomarker levels of MMP3 and CXCL13 in a separate cohort of patients with IPF (N=80).
Results 2940 genes were significantly differentially expressed between IPF and control samples (|fold change| >1.5, p<0.05). Two clusters of co-regulated genes related to bronchiolar epithelium or lymphoid aggregates exhibited substantial heterogeneity within the IPF population. Gene expression in bronchiolar and lymphoid clusters corresponded to the extent of bronchiolisation and lymphoid aggregates determined by immunohistochemistry in adjacent tissue sections. Elevated serum levels of MMP3, encoded in the bronchiolar cluster, and CXCL13, encoded in the lymphoid cluster, corresponded to disease severity and shortened survival time (p<10−7 for MMP3 and p<10−5 for CXCL13; Cox proportional hazards model).
Conclusions Microscopic pathological heterogeneity in IPF lung tissue corresponds to specific gene expression patterns related to bronchiolisation and lymphoid aggregates. MMP3 and CXCL13 are systemic biomarkers that reflect the aggregate burden of these pathological features across total lung tissue. These biomarkers may have clinical utility as prognostic and/or surrogate biomarkers of disease activity in interventional studies in IPF.
- Idiopathic pulmonary fibrosis
Statistics from Altmetric.com
Key messages
What is the key question?
-
How is gene expression in idiopathic pulmonary fibrosis (IPF) lung tissue linked to pathology, systemic biomarkers and clinical manifestations of disease?
What is the bottom line?
-
Using transcriptomic and pathological approaches, we identified two distinct features of IPF lesions: bronchiolisation and lymphoid aggregates; systemic levels of protein biomarkers encoded in these respective gene signatures (MMP3 and CXCL13) correspond to disease severity and survival in patients with IPF.
Why read on?
-
The unique systematic approach to understanding a complex human disease in this study sheds light on IPF disease progression and is generalisable to many indications.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive and fatal fibrotic lung disease with no known cause and a median survival of ∼3 years.1 The clinical course within the population of patients with IPF is variable, ranging from a slow, steady loss of lung function over 5 or more years to rapid progression and death within 1–2 years of diagnosis to relatively stable disease punctuated by intermittent precipitous declines in lung function resulting from acute exacerbations.2 The mechanisms underlying this variability in disease progression within the population of patients with IPF are poorly understood.
The fibrosis observed in IPF is hypothesised to originate from an irregular wound healing response triggered by a loss of integrity in the alveolar epithelium followed by persistent profibrotic signals that fail to resolve.3 The microscopic presentation of IPF in diseased lung tissue is spatially and temporally heterogeneous. Regions of fibroblast accumulation and active matrix remodelling can exist between areas of normal-appearing lung and mature scar tissue.4 A complex mixture of resident and infiltrating cell types can be found in these distinct regions of remodelled lung. It is therefore important to develop a deeper understanding of IPF biology at a local level to better characterise the location and consequences of aberrant signalling processes.
A challenge in designing interventional clinical studies in IPF is the lack of a robust means of identifying and controlling for biological heterogeneity, and of selecting patients at risk for outcomes of interest. Non-invasive biomarkers that reflect the activity of specific pathways and aggregate fibrotic burden across a given patient's total lung tissue could be valuable tools in identifying stage of disease, and could aid in selecting treatments and monitoring biological responses to therapy. In this study we used gene expression profiling and histology to characterise pathological patterns that display heterogeneous expression levels within lung tissue from a population of patients with IPF, and identified systemic biomarkers related to these patterns that correspond to disease severity. The coordinated analyses of gene expression, histology and peripheral biomarkers provide the opportunity to integrate insight into IPF pathogenesis at multiple levels, with the potential to contribute to the development of novel therapies and diagnostics.
Methods
Tissues were obtained from clinical samples from patients with IPF at the time of biopsy or lung transplantation. Peripheral blood and clinical data were obtained from a separate cohort of patients with IPF at the time of initial presentation to the interstitial lung disease clinic. All patients were seen at University of California, San Francisco (UCSF) and the diagnosis of IPF was established through multidisciplinary review of clinical, radiological and pathological data according to consensus criteria.5 Non-diseased normal lung tissues were procured from lungs not used by the Northern California Transplant Donor Network. Clinical and demographic information for the IPF cohorts is in online supplementary table S1 and table 1. Sample and data collection were approved by the UCSF Committee on Human Research and all patients provided written informed consent.
Clinical and demographic features of patients with idiopathic pulmonary fibrosis in the prognostic cohort (N=80)
Statistical inference analyses were performed in R. Relationship of expression signature scores or serum protein concentration to patient diagnosis was tested by the Wilcoxon rank-sum method. Relationship of expression signatures scores to histology scores was tested by a one-term logistic regression model. Correlations between MMP3 or CXCL13 and other variables were performed by the Spearman method. Survival analyses were performed using the Cox proportional hazards model with serum concentration as the only independent variable term and p values determined by the score test. Transplants were treated as censored events.
Detailed methods for RNA preparation, gene expression, immunohistochemistry (IHC) and biomarker analyses are available in the online data repository.
Results
Differential gene expression in IPF lung tissue
We performed genome-wide transcriptomic analysis of lung biopsy tissue from 40 patients with IPF and 8 control subjects without IPF (see online supplementary table S1). Eleven IPF samples were from video-assisted thoracoscopic (VATS) biopsies and 29 were from explants taken at the time of lung transplantation. Control tissue was explanted from unused donor lungs. RNA isolated from these samples was analysed using microarrays. Limited available metadata including sex, tissue source (VATS or explant) and diagnosis were included in a linear model used to identify differentially expressed (DE) genes. We identified 2940 probes as DE: 1531 upregulated and 1409 downregulated in IPF tissues compared with control tissues (fold-change >|1.5|, Benjamini-Hochberg adjusted p value <0.05; see online supplementary table S3). Among the significantly upregulated genes in IPF were those encoding multiple matrix metalloproteinases, collagens, and cytokines and growth factors. Many DE genes in IPF lung tissue overlapped with those reported in previously published data sets.6–8
Heterogeneity in clusters of coregulated genes
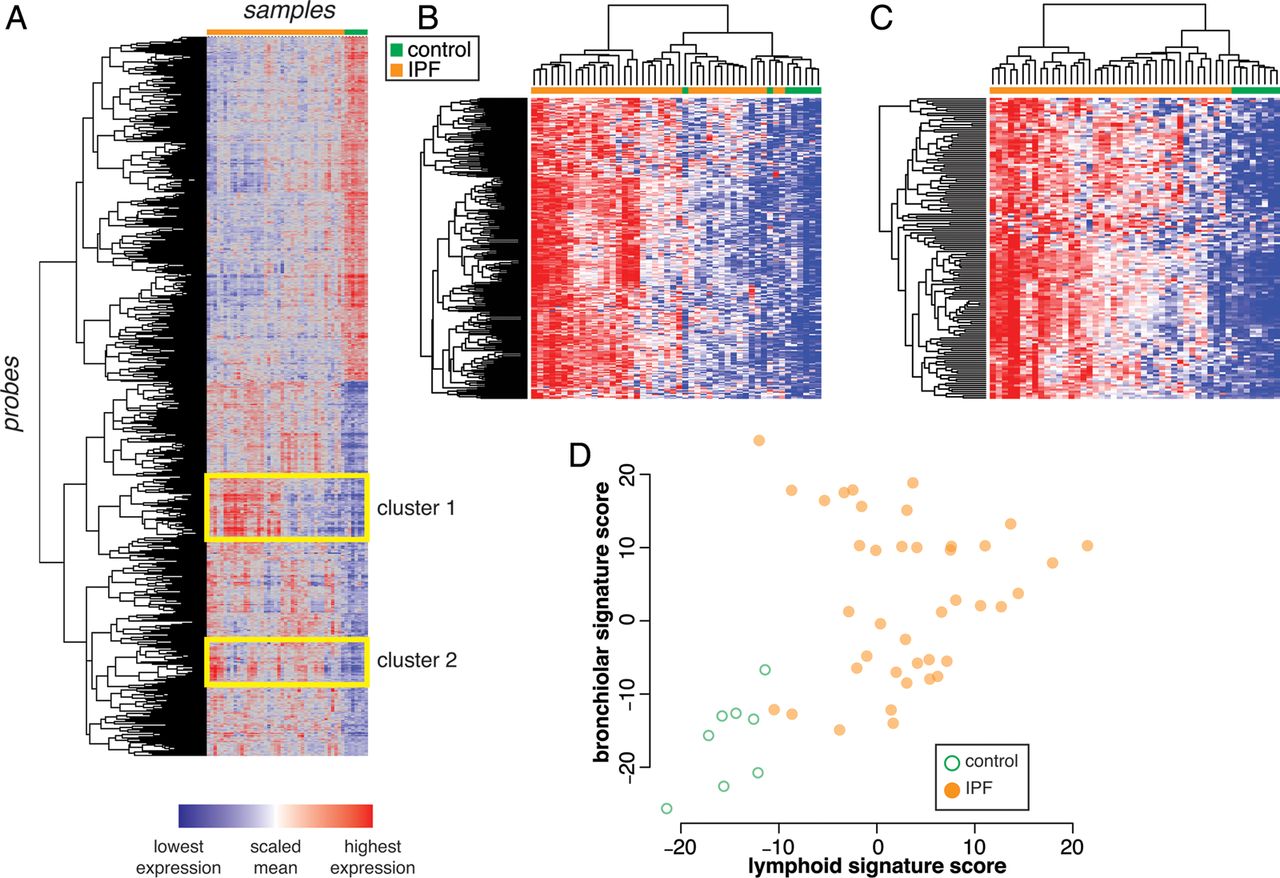
Unsupervised 2-way hierarchical clustering of the 2490 DE probes between IPF and control demonstrated two major clusters of samples defined by diagnosis (figure 1A). Inspection of the dendrogram revealed two clusters of coregulated genes that were upregulated in IPF compared with controls, yet were variable within the IPF population.

Heterogeneous clusters of coexpressed genes among idiopathic pulmonary fibrosis (IPF) lung biopsy samples. (A) Unsupervised 2-way clustering of all differentially expressed genes (|fold change| >1.5; Benjamini-Hochberg adjusted p value <0.05) between IPF (N=40) and control (N=8) lung biopsy samples. IPF samples are indicated in orange and control samples are indicated in green. (B) Reclustering of samples on coregulated genes in cluster 1 (‘bronchiolar’ cluster). (C) Reclustering of samples on coregulated genes in cluster 2 (‘lymphoid’ cluster). (D) Normalised mean expression scores for all genes in the bronchiolar and lymphoid clusters.
Cluster 1 comprises genes related to bronchiolar epithelium, including mucins (MUCL1, MUC4, MUC20), proline-rich secreted factors (PRR7, PRR15, SPRR1B, SPRR2D), keratins (KRT5, 6B, 13, 14, 15, 17), serine protease inhibitors (SERPINB3, B4, B5, B13), ion channels and associated factors (TRPV4, CLCA2), and cilium components (BBS5). This is consistent with prior reports of abnormal ‘bronchiolisation’ of alveolar spaces in IPF, which may represent epithelialisation of honeycombed cystic spaces in regions of dense scarring,9–11 hence we have termed this cluster of coregulated genes the ‘bronchiolar signature.’ A full list of genes in the bronchiolar signature is in online supplementary table S4. Reclustering the samples using only the bronchiolar signature genes emphasises the range in gene expression levels observed within the IPF population (figure 1B).
Cluster 2 consists of T cell and B cell markers (CD19, CD20, CD27, CD28), Fc receptor genes (FCRLA, FCRL2, FCRL5), and chemokines and chemokine receptors (CXCL13, CXCR5, CCR6, CCR7). This pattern is consistent with prior reports showing ectopic lymphoid tissue in regions of established fibrosis in biopsies from IPF,12–14 hence we have termed this cluster of coregulated genes the ‘lymphoid signature.’ A full list of genes in this cluster is in online supplementary table S5. Reclustering the samples using only the lymphoid gene signature genes shows heterogeneity across the IPF samples (figure 1C).
To determine whether the bronchiolar and lymphoid signatures were related to each other in individual samples, we derived summary gene expression scores for each signature by calculating the normalised mean expression of all the genes in each cluster. While samples from most patients with IPF had higher bronchiolar or lymphoid signature scores than controls, there was substantial variability within the IPF population. While nearly all samples with markedly elevated lymphoid signatures also had elevated bronchiolar signatures, the converse was not true (figure 1D). These results suggest that the signatures may reflect distinct biological processes heterogeneously expressed across patients with IPF and/or differential sampling of processes that have heterogeneous spatial distribution within bulk IPF lung tissue.
Localisation of signature markers to specific histological features in IPF lung tissue
IPF lung tissue is histologically heterogeneous on a microscopic scale as evidenced by a low magnification H&E image (figure 2A). Higher magnification of selected regions reveals relatively normal alveolar tissue (figure 2B) adjacent to ‘transition zones’ of active fibrogenesis with thickened alveolar walls (figure 2C) and advanced scar tissue with regions of dense fibrosis interspersed with honeycombed cysts lined with columnar epithelium (figure 2D).
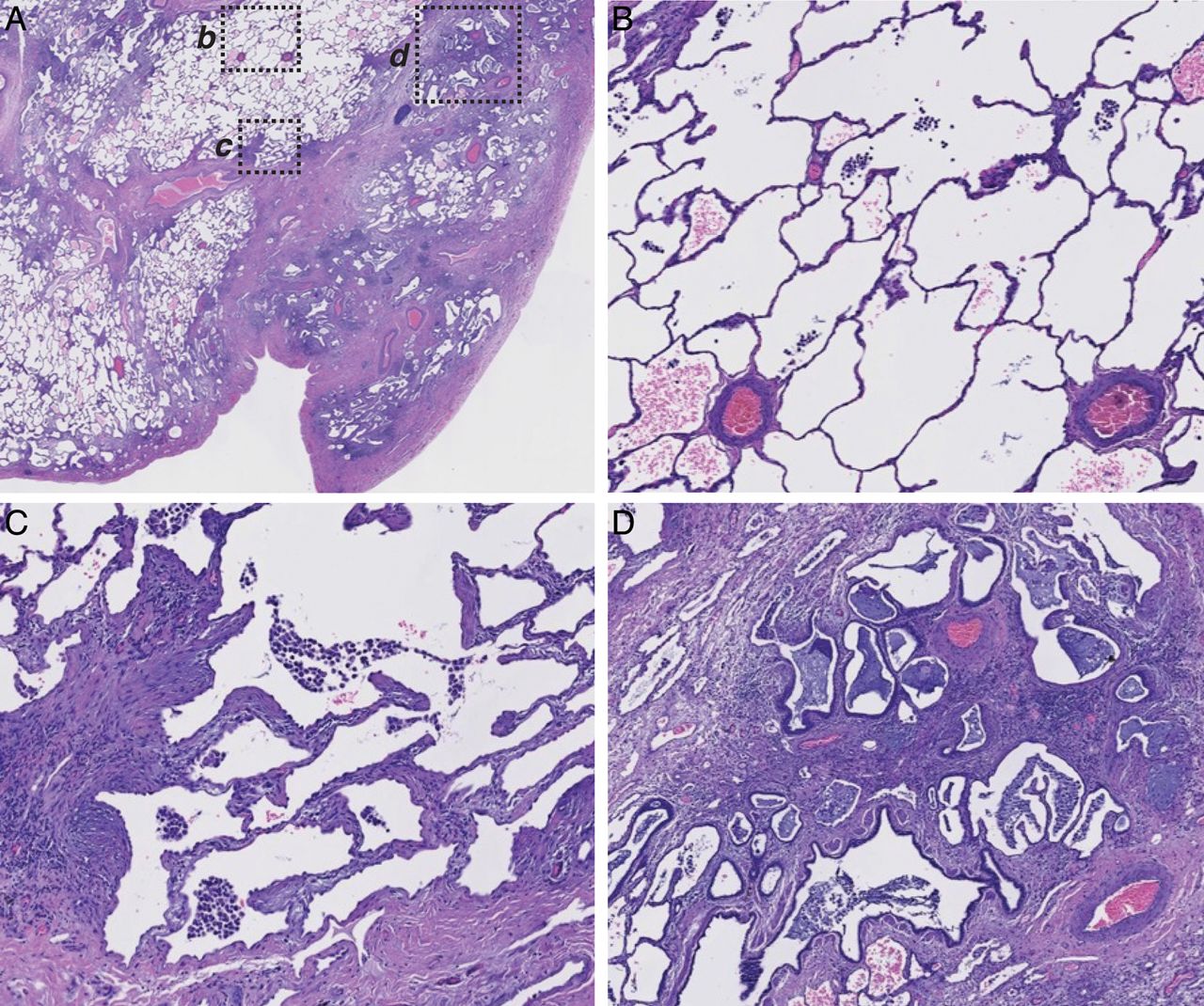
Spatial heterogeneity of pathophysiology in a single representative idiopathic pulmonary fibrosis (IPF) biopsy. (A) Low-power magnification of a H&E-stained paraffin section of explanted lung tissue from a patient with IPF. Pleural surface is at lower right. b, c, and d indicate regions shown at higher magnification in: (B), normal-appearing alveolar structure, (C) transition zone underlying normal-appearing parenchyma with thickened alveolar walls, (D) advanced scar tissue with microscopic honeycombing and bronchiolisation.
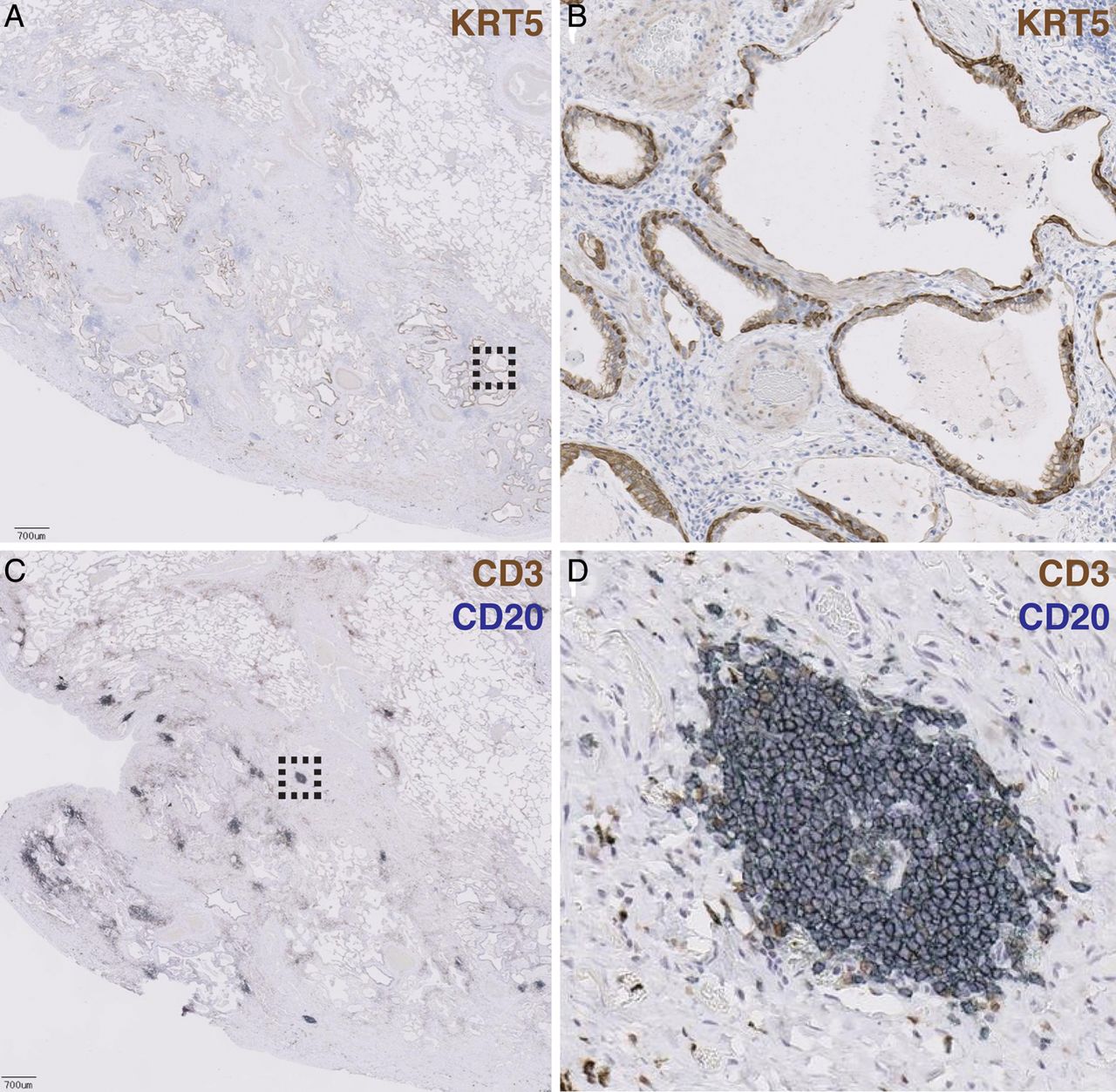
To determine where selected genes encoded in each of the signatures were spatially distributed in regions of lung tissue, we performed IHC on biopsy tissue taken from IPF lung explants (N=10). Cystic structures were lined with epithelial cells expressing keratin 5, a gene encoded in the bronchiolar signature (figure 3A, B). H&E, trichrome, anti-KRT14 and periodic acid-Schiff staining of adjacent sections further revealed that these structures were lined with columnar epithelial cells with abundant cytoplasm, near to but distinct from regions of collagen deposition, with coincident expression of mucins (see online supplementary figure 1A–D). Taken together, these patterns are consistent with cystic spaces in IPF lung lined with bronchiolar epithelium.

Bronchiolisation and lymphoid aggregates occur in spatially distinct regions of advanced scar tissue in idiopathic pulmonary fibrosis (IPF) biopsies. (A) Low-power magnification of an anti-keratin 5 (KRT5)-stained paraffin section of explanted lung tissue from a patient with IPF. Pleural surface is at lower left. Scale bar=700 μM. Dashed box indicates region shown at higher magnification in panel (B). (C) Adjacent section costained with anti-CD3 (brown) and anti-CD20 (blue) showing lymphoid aggregates. Dashed box indicates region shown at higher magnification in panel (D).
In spatially discrete areas from bronchiolised regions, we observed numerous aggregates of CD20+ B lymphocytes and CD3+ T lymphocytes (figure 3C, D). H&E and trichrome staining revealed clusters of cells with darkly staining nuclei, near to but distinct from regions of high collagen deposition; these cells stained positively for CD20 (see online supplementary figure S1E–H). Taken together, these patterns are consistent with lymphoid aggregates.
Gene expression signatures correlate to specific histological features in IPF tissue
To confirm that the gene signatures observed in the microarray experiment corresponded to bronchiolar and lymphoid structures observed histologically in IPF lung tissue, we performed a coordinated analysis of gene expression and histological evaluation on serial tissue sections from lung biopsies, schematically depicted in figure 4A.

Bronchiolar and lymphoid gene signature expression scores correspond to extent of bronchiolisation and lymphoid aggregates in adjacent tissue sections. (A) Schematic depiction of method to compare histopathology and gene expression in adjacent tissue sections. (B) qPCR-determined bronchiolar gene signature score in control and idiopathic pulmonary fibrosis (IPF) tissue scrapings. (C) qPCR-determined lymphoid gene signature score in control and IPF tissue scrapings. *p Values for (B) and (C) determined by Wilcoxon rank-sum method. (D) qPCR bronchiolar gene signature score as a function of bronchiolar histology score. (E) qPCR lymphoid gene signature score as a function of lymphoid histology score. **p Values for (D) and (E) determined by logistic regression (IPF samples only; controls excluded from statistical analysis).
We isolated total RNA from tissue sections and assessed gene expression by qPCR using 24 probes spanning the bronchiolar and lymphoid signatures (see online supplementary table S2) and derived expression indices for each signature from the qPCR data by taking the normalised mean expression of all the genes in each cluster across all samples (centered at 0). We first tested the qPCR platform with the same RNA isolated from bulk lung biopsies used in the microarray analyses of gene expression described above and observed highly concordant signature scores between the microarray and qPCR experiments (see online supplementary figure S2) irrespective of tissue source (VATS biopsy or explant). Gene expression in individual tissue sections was significantly enriched in IPF compared with control lung tissue for the bronchiolar and lymphoid signature scores (figure 4B, C). Sections adjacent to those assessed by qPCR were assessed by IHC and scored by a pathologist blinded to the expression scores for the presence of bronchiolisation and lymphoid aggregates on a 0–3 scale, with ‘0’ representing the absence of a particular feature and ‘3’ representing a section that displays that feature prominently (examples in online supplementary figure S3). There were highly significant correlations between histology and gene expression scores for the bronchiolar and lymphoid signatures (figure 4D, E). Taken together, these data further substantiate that the gene expression patterns observed by microarray correspond to specific and distinct pathological features of IPF lesions.
Signature genes encoding candidate biomarkers in IPF
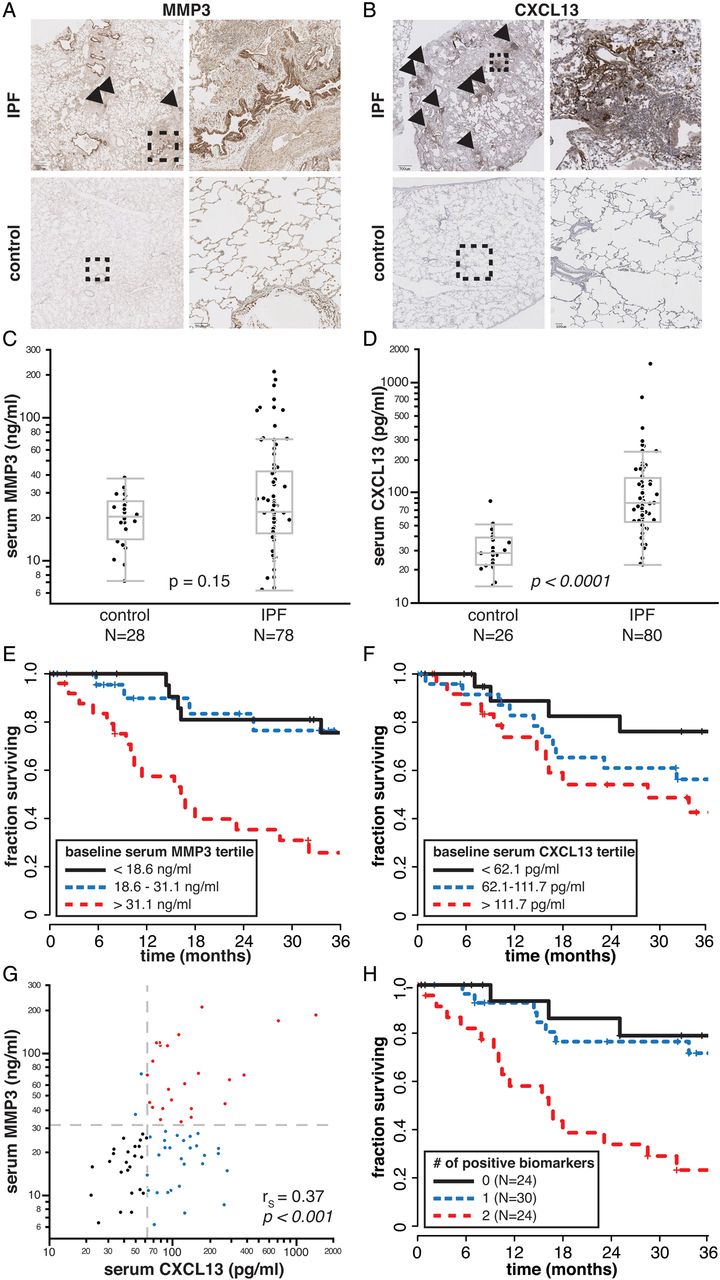
As bronchiolisation and lymphoid aggregates are heterogeneously distributed within individual IPF biopsies (figure 3), a small biopsy sample may not accurately represent the aggregate pathological burden across the total lung tissue in a given patient with IPF. However, soluble secreted factors produced by those pathological structures may be detectable in peripheral blood and have the potential to reflect the total pathological burden within a patient. Therefore, we identified genes within the bronchiolar and lymphoid signatures that encode soluble proteins known to be detectable in peripheral blood to investigate in peripheral blood as biomarker candidates that may provide insight into clinical presentation and disease progression in IPF. MMP3 is a matrix metalloproteinase encoded in the bronchiolar signature and CXCL13 is a chemokine encoded in the lymphoid signature; both are soluble secreted factors that are detectable in peripheral blood.15 ,16 IHC revealed strong staining for MMP3 predominantly in epithelial cells lining bronchiolised regions in dense scar tissue (figure 5A), with modest immunoreactivity evident in some lymphoid aggregates. CXCL13 protein stained preferentially in the periphery of lymphoid aggregates and was not evident in bronchiolised regions (figure 5B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Elevated serum MMP3 and CXCL13 levels are prognostic for decreased survival time in patients with idiopathic pulmonary fibrosis (IPF). (A) Immunohistochemistry (IHC) for MMP3. Dashed boxes on left panels indicate region shown at higher magnification in right panels. Arrowheads indicate lymphoid aggregates. Scale bar=700 μM (upper left panel); 90 μM (lower right). (B) IHC for CXCL13 as in (A). Scale bar=700 μM (upper left); 100 μM (lower right). (C) Serum MMP3 levels in controls and patients with IPF. (D) Serum CXCL13 levels in controls and patients with IPF. p-values for (C) and (D) determined by the Wilcoxon rank-sum method. (E) Kaplan-Meier (K-M) survival plot for patients with IPF by tertiles of baseline serum MMP3 levels. (F) K-M survival plot for patients with IPF by tertiles of baseline serum CXCL13 levels. (G) Correlation between serum MMP3 and CXCL13 levels (patients with IPF only). rS, Spearman's rank-order correlation coefficient. (H) K-M survival plot for patients with IPF by combined baseline serum MMP3 and CXCL13 levels. A score of 0 corresponds to MMP3 <31.1 ng/mL and CXCL13 <62.1 pg/mL; a score of 1 corresponds to either MMP3 >31.1 ng/mL or CXCL13 >62.1 pg/mL; a score of 2 corresponds to MMP3 >31.1 ng/mL and CXCL13 >62.1 pg/mL.
We evaluated serum levels of MMP3 and CXCL13 in a separate cohort of 80 patients with IPF collected at the time of initial presentation to the ILD clinic (table 1), and 28 healthy controls (not age-matched or sex-matched; average age 40 years, range 26–64 years old, 56% male). The distribution of serum MMP3 mostly overlapped between controls and patients with IPF; however approximately a third of patients with IPF had elevated serum MMP3 levels compared with controls (figure 5C). Serum CXCL13 was significantly elevated in patients with IPF compared with controls (figure 5D).
Among patients with IPF, serum MMP3 levels were positively correlated with dyspnoea score and negatively correlated with FVC, while serum CXCL13 levels were positively correlated with dyspnoea score and negatively correlated with diffusing capacity of carbon monoxide (DLCO) (table 2). Both biomarkers were significantly negatively correlated with survival over 3 years subsequent to blood sampling, as assessed using continuous correlations and a Cox proportional hazards model (table 2). These effects remain significant when the model is adjusted for baseline FVC, DLCO, sex and age (see online supplementary table S6). We extended the analysis of this relationship via Kaplan–Meier plots of survival by tertiles of serum biomarkers, which suggested that patients with IPF in the top tertile of baseline serum MMP3 or the top two tertiles of baseline serum CXCL13 levels (figure 5E, F) had shortened survival times. Combinations of biomarkers with thresholds of 31 ng/mL MMP3 (top tertile) and 62 pg/mL CXCL13 (top 2 tertiles) demonstrated that patients with elevated levels of both biomarkers had substantially shorter survival times than patients with elevated levels of neither or only one biomarker. However, most of this effect is due to MMP3, as MMP3 and CXCL13 levels were positively correlated in patients with IPF; nearly all patients with elevated MMP3 also had elevated CXCL13, but not all patients with elevated CXCL13 had elevated MMP3 (figure 5G, H). Taken together, these data show that blood biomarkers encoded by genes expressed in pathological structures associated with advanced lesions in IPF lung tissue are correlated with clinical disease severity in patients with IPF.
Correlations between blood biomarkers, lung function and survival among patients with idiopathic pulmonary fibrosis
Discussion
The analysis of gene expression in lung tissue from patients with IPF can provide insights into aberrant molecular and cellular processes associated with disease pathology. However, in cross-sectional analyses of bulk biopsy tissue, it is difficult to discern when, where and how particular sets of DE genes exert their effect. We have taken complementary approaches, coordinating gene expression with pathological analyses of lung tissue in focal biopsies and extending those findings to peripheral biomarkers and clinical data. Systemic biomarkers that reflect focal pathological processes in diseased lung have the potential to integrate the cumulative burden of these processes across the entirety of patients’ lung tissue and provide insights into clinically relevant disease features.
We identified two clusters of coregulated genes in IPF lung tissue that display significant heterogeneity across the sample cohort. The genes in these clusters correspond to bronchiolar epithelium and lymphoid aggregates, respectively. Pathological examination of IPF biopsies reveals ciliated columnar epithelial cells lining cystic structures and clusters of lymphocytes in regions of advanced scarring. Elevated expression of genes encoded in the respective signatures was correlated with the relative prevalence of those features in a given tissue section.
Pathological implications of the bronchiolar and lymphoid gene signatures
Bronchiolisation of alveolar tissue is a well-described feature of the lung remodelling in IPF lesions.9–11 We observed keratin-expressing columnar cells lining cystic structures in regions of advanced scarring near the pleural surfaces, consistent with the structures being lined with bronchiolar epithelial cells. Whether the columnar epithelial cells lining honeycomb cysts arise from adjacent normal bronchiolar structures or de novo bronchiolisation of alveolar tissue remains uncertain, but their presence is a feature of advanced fibrosis and honeycomb cysts found in IPF lung tissue.
MMP3 clusters with genes in the bronchiolar signature (see online supplementary table S4) and exhibits strong immunoreactivity in bronchiolised regions of IPF lung biopsy tissue (figure 5A). MMP3 can degrade extracellular matrix components, and its activity has been implicated in wound repair and tumour initiation.17 ,18 Elevated levels of MMP3 gene and protein expression have been described in IPF lung tissue, with MMP3 expression localised to alveolar and bronchiolar epithelial cells in IPF but not control lung tissue.19 Elevated MMP3 protein levels have been reported in bronchoalveolar lavage fluid from patients with IPF.20 Ectopic overexpression of MMP3 in rat lungs promoted pulmonary fibrosis while MMP3-deficient mice were protected from bleomycin-induced pulmonary fibrosis relative to wild type mice.19 Hence MMP3 may play a role in the chronic pathological wound repair and tissue remodelling characteristic of advanced IPF lesions.
The bronchiolar signature is similar to a set of genes reported to be upregulated in lung tissue from patients with concomitant pulmonary fibrosis and pulmonary artery hypertension (PAH).21 Hence it is possible that the decreased tissue compliance and impaired gas exchange associated with advanced scarring, honeycombing and bronchiolisation is associated with increased pulmonary vascular resistance and PAH. PAH in patients with IPF is associated with dramatically reduced survival time.22 ,23 Unfortunately, we lacked data on whether patients in our cohort had PAH, but we hypothesise that elevated serum MMP3 levels may be indicative of PAH in patients with IPF, which should be formally assessed in future studies.
The ‘lymphoid’ signature suggests an inflammatory component associated with chronic lesions in IPF,24 although the exact nature of immune cell aggregates’ role in IPF pathology is unclear. Most T cells and B cells present in IPF reside in loose focal aggregates as determined by CD3 and CD20 immunolocalisation. CXCL13 is a chemokine produced by follicular dendritic cells. It recruits B cells to secondary and tertiary lymphoid structures by binding to its cognate receptor CXCR5, and CXCL13 and CXCR5 are required for lymphoid follicle formation.25 Lymphoid aggregates have been reported to associate with usual interstitial pneumonia lesions in IPF biopsies12 and B cell infiltrates with high CXCL13 expression have also been reported in COPD.26–28 Serum CXCL13 is a biomarker of the severity of joint erosions29 and of B cell repopulation after rituximab treatment in rheumatoid arthritis16; a recent report has described increased CXCL13 expression in IPF lungs and plasma CXCL13 as prognostic in an independent cohort of patients with IPF.14 In that study, increases in plasma CXCL13 levels over time corresponded to an increased rate of respiratory failure. This trend is consistent with CXCL13 being a biomarker of advanced disease in IPF and suggests that lymphoid aggregates are a late manifestation of the disease.
Whether lymphoid aggregates in IPF are a cause or a consequence of other molecular and cellular processes is presently unclear. Some studies have suggested an autoimmune component to IPF, with increased incidence of autoantibodies against periplakin30 and Hsp70,31 and increased levels of B lymphocyte stimulator, a B cell survival factor,32 in patients with IPF, each of which was associated with increased disease severity and poor prognosis. Other studies have reported similar prevalence of common autoantibodies in patients with IPF and healthy age-matched controls.33 No studies to date have reported whether therapeutic interventions specifically targeting lymphoid biology have any therapeutic benefit in IPF. In our study, lymphoid aggregates were generally found in regions of dense scar tissue, and exhibited immunoreactivity for CXCL13 and MMP3, which could explain the moderate correlations between bronchiolar and lymphoid signatures and MMP3 and CXCL13 levels in peripheral blood. Bronchiolisation and lymphoid neogenesis may be common processes downstream of heavy fibrotic burden within IPF lungs. The prognostic properties of CXCL13 presented here, taken together with prior reports, further substantiate the significance of lymphoid aggregates and adaptive immune responses with respect to disease severity and prognosis in IPF.
Conclusion: potential applications of IPF biomarkers
We have shown that biomarkers encoded in gene expression signatures correlated with distinct pathological structures in IPF lung tissue are detectable at elevated, but variable, levels in the peripheral blood of patients with IPF. MMP3 and CXCL13 levels in peripheral blood are associated with features of disease severity, including lung function, diffusing capacity, dyspnoea and survival time. Given the geographical heterogeneity and patchy nature of IPF pathology, systemic biomarkers may provide a more comprehensive picture of the aggregate burden of specific pathological disease features in an individual patient with IPF than gene expression levels from or histological examination of a focal biopsy. A potential confounder in this study is that a quarter of the patients in the biomarker cohort were taking systemic immunosuppressants (steroids and/or azathioprine) at the time of sample collection, which could potentially affect systemic biomarker levels and/or survival time. In light of evolving IPF clinical management strategies including recommendations against systemic steroid treatment34 and new therapies such as pirfenidone and nintedanib,35 future work should be directed at replicating these findings and assessing the dynamics of MMP3 and CXCL13 in additional cohorts of patients with IPF over time with respect to treatment and clinical disease progression, and exploring whether these biomarkers can be incorporated along with clinical and physiological variables into tools that predict rates of disease progression and mortality.36
Acknowledgments
The authors thank the patients who participated in this study and the providers who referred these patients to the UCSF ILD clinic. Without the generous donation of biological samples from patients, this study could not have been performed.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
DJD and SC contributed equally.
-
PJW and JRA codirected this project.
-
Contributors Conception and design: DJD, SC, HRC, JGE, PJW and JRA; Analysis and interpretation: DJD, SC, ARA, GJ, ENN, PC, SEK, SB, SKK, CH, ZM, MAM, JK, HRC, JGE, PJW, JRA; Drafting the manuscript for important intellectual content: DJD, SC, HRC, JGE, PJW and JRA.
-
Funding Genentech, the Nina Ireland Lung Disease programme at UCSF, and NIH grant HL 108794.
-
Competing interests DJD, ARA, GJ, ENN, PC, SEK, SB, CH, MAM, JGE and JRA are employees of Genentech and may hold stock or stock options in the Roche Group. SC and SKK are former employees of Genentech.
-
Ethics approval UCSF Committee on Human Research.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Microarray data are available from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/; GEO accession ID GSE53845).