Article Text

Abstract
Background Complex polymicrobial communities infect cystic fibrosis (CF) lower airways. Generally, communities with low diversity, dominated by classical CF pathogens, associate with worsened patient status at sample collection. However, it is not known if the microbiome can predict future outcomes. We sought to determine if the microbiome could be adapted as a biomarker for patient prognostication.
Methods We retrospectively assessed prospectively collected sputum from a cohort of 104 individuals aged 18–22 to determine factors associated with progression to early end-stage lung disease (eESLD; death/transplantation <25 years) and rapid pulmonary function decline (>−3%/year FEV1 over the ensuing 5 years). Illumina MiSeq paired-end sequencing of the V3-V4 region of the 16S rRNA was used to define the airway microbiome.
Results Based on the primary outcome analysed, 17 individuals (16%) subsequently progressed to eESLD. They were more likely to have sputum with low alpha diversity, dominated by specific pathogens including Pseudomonas. Communities with abundant Streptococcus were observed to be protective. Microbial communities clustered together by baseline lung disease stage and subsequent progression to eESLD. Multivariable analysis identified baseline lung function and alpha diversity as independent predictors of eESLD. For the secondary outcomes, 58 and 47 patients were classified as rapid progressors based on absolute and relative definitions of lung function decline, respectively. Patients with low alpha diversity were similarly more likely to be classified as experiencing rapid lung function decline over the ensuing 5 years when adjusted for baseline lung function.
Conclusions We observed that the diversity of microbial communities in CF airways is predictive of progression to eESLD and disproportionate lung function decline and may therefore represent a novel biomarker.
- cystic fibrosis
- respiratory infection
Statistics from Altmetric.com
Key messages
What is the key question?
Cross-sectional studies have demonstrated that the complexity of microbial communities in sputum from individuals with cystic fibrosis (CF) inversely correlates with baseline disease state.
However, longitudinal studies evaluating the microbiota as a novel biomarker in predicting future CF lung disease progression are lacking.
What is the bottom line?
Our group assessed, for the first time, the risk of progression to early end-stage lung disease and accelerated lung function decline in a cohort of 104 young adults with CF who had sputum samples collected 5 years previously.
Whereas traditional cultured pathogens did not correlate with subsequent outcomes, a microbial community of limited diversity, dominated by Pseudomonas, independently predicted a negative prognosis.
Why read on?
This study suggests that microbial community analysis may serve a novel biomarker to identify CF individuals at the highest risk of disease progression.
Introduction
Progressive respiratory infection is responsible for the majority of cystic fibrosis (CF) morbidity and mortality.1 Traditional sputum cultivation identifies a range of airway pathogens. However, cultured pathogens do not adequately distinguish individuals at increased risk of deterioration.2–4 Accordingly, the observation that a polymicrobial community of organisms overlooked by traditional protocols colonising CF airways has proved tantalising.5 6
Cross-sectional studies have identified that sputum from patients with advanced disease have limited diversity and are dominated by organisms such as Pseudomonas aeruginosa, Stenotrophomonas maltophilia and Burkholderia cepacia complex (Bcc).7 8 However, few longitudinal studies have attempted to correlate the CF microbiome with future clinical outcomes.6 We hypothesised that the airway microbiota of a cohort of adolescents with CF would associate with subsequent long-term clinical outcomes and serve as a novel biomarker. Microbial community analysis could then serve as a new clinical tool to identify patients at increased risk of disease progression.9 10
Methods
Patients and sample selection
The Calgary Adult CF Clinic Sputum Biobank includes prospectively collected sputum samples from 1998 to 2017 stored at −80°C using conserved quality control storage strategies previously described.11 For this study, we included patients with sputum collected between the ages 18 and 22. To ensure that samples were collected under similar conditions we excluded samples collected within 28 days of changes in therapy or pulmonary exacerbation (PEx). We captured patient characteristics, including: demographics, chronic therapies, semiquantitative cultured pathogens, percent predicted FVC and FEV1 at the time of sample collection. Patients were categorised as having mild (FEV1: >80%), moderate (FEV1: 40%–80%) and advanced lung disease (FEV1: <40%) at baseline.11 Disease progression over the ensuing 5 years was assessed through detailed record audits. Rates of lung function decline were determined through subject-specific constructed linear regressions. As the study included patients and samples spanning two decades, patients were categorised into three cohorts based on the years in which they transitioned into the adult clinic: cohort_A (1998–2003), cohort_B (2004–2008) and cohort_C (2009–2013).
Clinical outcomes
The primary outcome was progression to early end-stage lung disease (eESLD) defined as death/transplantation <25 years—a value chosen based on disproportionate disease progression.12–14 To determine factors associated with eESLD, patients meeting this definition were compared with those who did not (nESLD). Our secondary outcome of interest was factors associated with accelerated lung function decline. Patients were categorised as either rapid progressors (RP) (ie, FEV1 decline worse than −3%/year) or non-rapid progressors (Nrp) based on decline over the ensuing 5 years. These values were chosen based on accelerated decline experienced by outliers.10 15–17 Factors associated with accelerated decline were assessed using both an absolute (Ab; annual FEV1% decline) and relative (Re; absolute rate/baseline) definition.
16S rRNA gene sequencing and processing
Total DNA extraction from frozen sputum samples and reagent blanks was performed as previously described.11 Bacterial communities in CF sputum and reagent blanks were characterised by amplification and sequencing the V3-V4 region of the 16S rRNA.11 The sequencing reads were processed and analysed to construct the operational taxonomic unit (OTU) table using a UPARSE pipeline.18 After removing singleton OTUs 8 320 948 total reads remained (average 80 009 reads/sample; IQR: 57 168–102 441).
Statistical analysis
Richness and evenness was analysed using Shannon’s and Simpson’s diversity indices. Richness estimators (Observed and Chao1) were calculated. After proportional normalisation of all samples, Bray-Curtis dissimilarity was calculated to analyse community beta diversity. Permutational multivariate analysis of variance (PERMANOVA) test19 was performed to determine the significance related to the beta-diversity analysis. Beta diversity was calculated after rarefying all samples to 15 000 reads. Non-metric multidimensional scaling plots were used to visualise Bray-Curtis dissimilarity matrixes. Fisher’s exact and Χ2 tests were used to discriminate between dichotomous variables. Wilcoxon rank-sum tests were conducted for continuous variables. A multivariable logistic regression model was constructed to predict the progression to eESLD. A forward stepwise selection process was used to select variables (those with p<0.2 in univariate analysis) independently associated with the primary outcome. Analyses were conducted with R V.3.2.1 (R Core Team, 2014) and STATA V.14.2 (StataCorp, Texas, USA).
Results
Study cohort
One hundred and thirty patients aged 18–22 were screened and 104 were included for analysis. Twenty-one were excluded because of inadequate follow-up (<5 years without reaching primary outcome) and five because all available samples were impacted by PEx and/or receipt of acute antibiotics. Patients who were excluded did not differ with respect to F508del homozygosity, pancreatic sufficiency or gender (p>0.05).
Traditional factors associating with clinical outcomes
Demographics, concomitant pathogens and treatments are presented in table 1. At sputum collection, patients were classified as having mild: 15 (14.4%), moderate: 44 (42.3%) and advanced: 45 (43.2%) lung disease. Of the cohort, 17 (16.3%) progressed to eESLD (5 died, 12 received life-saving lung transplantation) at a median age of 21.7 years (IQR 19.8–23.7). Patients progressing to eESLD relative to nESLD had worse baseline lung disease and nutrition (table 1), and were more likely to be receiving inhaled antipseudomonals, DNase and enteral/nutritional supplements (DNS; data not shown). Notably, neither cultured CF pathogens (including P. aeruginosa assessed as mucoid) nor the log level at which they were cultured (DNS) associated with progression to eESLD or RP status (table 1). Rates of progression to eESLD did not differ based on transition cohort (A: 22%; B: 18%; C: 9%, p=0.374).
Baseline characteristics of the study population as a function of the clinical outcomes
Accelerated lung function decline was observed in 58 patients (55.7%) when measured as an absolute FEV1 decline >−3%/year. Demographic, treatment or cultured pathogen factors were not associated with rate of absolute lung function (table 1, DNS). Similar results were obtained when assessed 2.5 years after collection (DNS). Forty-seven patients (45.1%) met the pre-determine definition of relative rapid progression (Re-RP) (table 1). Out of the 17 patients who were categorised as eESLD, 13 (76%) were categorised as either Ab-RP or Re-RP.
Factors associated with accelerated relative FEV1 decline included baseline lung function, nutritional status and osteoporosis/osteopenia (table 1). Microbial factors (including P. aeruginosa mucoid) did not associate with accelerated relative FEV1 decline. Categorisation as Ab-RP did not differ based on transition cohort (A: 51.8%; B: 68.1%; C: 42.4%, p=0.075), but did as Re-RP (A: 37%; B: 59%; C: 33.3%, p=0.05), although older cohorts were not disproportionally affected.
CF microbial community analysis
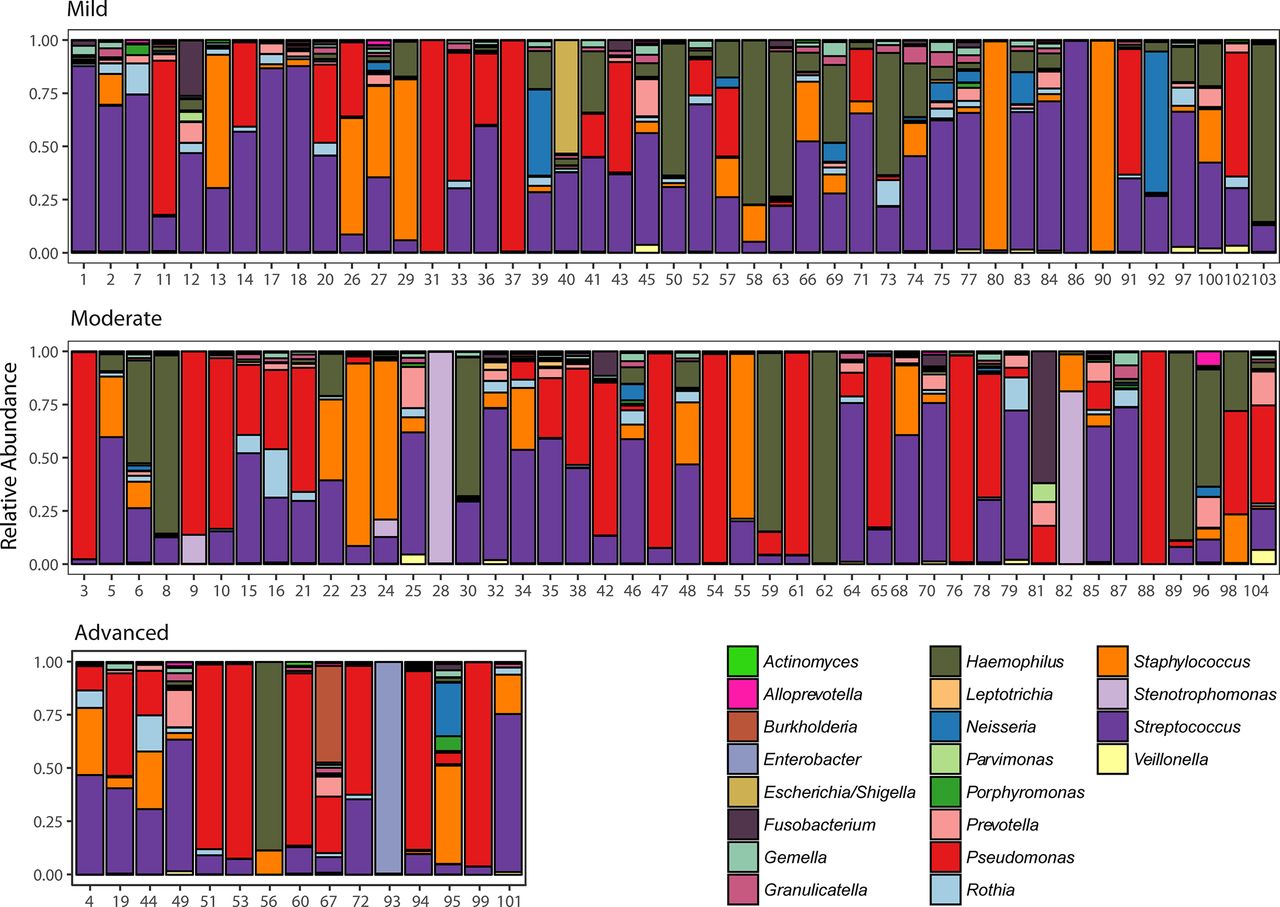
To visualise airway community composition we plotted the relative abundance (RA), at genus level, as a function of patient’s baseline FEV1 (figure 1). Genera classified as Pseudomonas, Streptococcus, Staphylococcus and Haemophilus had the greatest RA, representing 80.7% of reads. To ensure a single sample was adequate, a subset of 35 patients (33.6%), who had two appropriate sputum samples collected a median 151 days apart (IQR 60–212), was further assessed for stability. Samples clustered by patient, explaining 75.9% of community variance (p=0.001, PERMANOVA) (online supplementary figure E1).
Supplementary file 1

Composition of the lower airway microbiota in patients with cystic fibrosis (CF). Relative abundance of sputum bacteria reported at the genus level from individuals aged 18–22 with mild (upper panel), moderate (middle panel) and advanced (lower panel) lung disease. The top 20 genera accounting each for at least >0.28% abundance of the whole data set are coloured and presented in the figure. Numbers denote patient ID.
Microbiota associated with the primary outcome: progression to eESLD
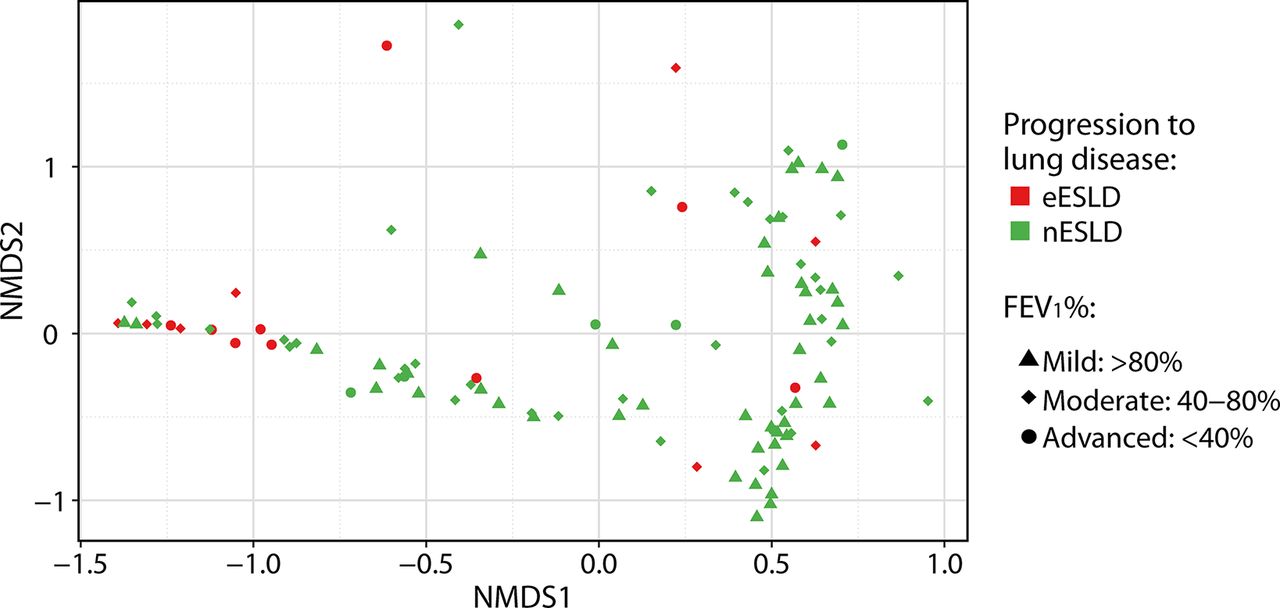
Sputum from patients who subsequently progressed to eESLD had a significantly lower alpha diversity compared with nESLD using multiple measures of richness and evenness (figure 2A). When we performed a sensitivity analysis using alternate definitions of eESLD (prior to 23 or 27 years), the same trends were evident (DNS). Comparisons of community structures revealed that sputum from eESLD had microbial communities that clustered after proportionally normalising all samples (p=0.003, PERMANOVA) (figure 3). Similar results were obtained when the analysis was performed with rarefied data (p=0.002, PERMANOVA). Furthermore, we observed that beta diversity correlated with baseline FEV1% using non-rarefied (p=0.032) and rarefied data (p=0.015, PERMANOVA).

Comparison of alpha-diversity indexes and primary and secondary outcomes. Box plot comparison of Observed, Chao1, Shannon and Simpson estimators for progression to eESLD (A), absolute rate of lung function (B) and relative rate of lung function (C). The median and IQRs are indicated as the middle, top and bottom lines of each box. Wilcoxon rank-sum test was performed. Ab, absolute; eESLD, early end-stage lung disease; nESLD, no eESLD; Nrp, non-rapid progressors; Re, relative; RP, rapid progressors.

NMDS plot of cystic fibrosis (CF) sputum from individuals aged 18–22. Each symbol represents one community from one patient. Beta-diversity plots based on Bray-Curtis dissimilarities from patients with progression to early end-stage lung disease (eESLD, red) or not (nESLD, green). Samples were shape coded by their baseline lung disease status at sample collection (ie, FEV1% predicted values: <40% (●), 40%–80% (♦) and >80% (▲)). Samples were separated according to subsequent progression to eESLD and baseline FEV1% status (p=0.003, R2=3.9% and p=0.006, R2=3.2%, respectively, permutational multivariate analysis of variance (PERMANOVA) test). NMDS, non-metric multidimensional scaling.
Given the typical co-occurrence of communities of limited diversity and Pseudomonas-dominated communities, we performed subgroup analyses to test if the association of eESLD still existed in patients without Pseudomonas domination. When we restricted our analysis to patients who were culture negative for P. aeruginosa (n=40), we observed that communities of patients who subsequently progressed to eESLD similarly trended towards lower alpha diversity (Observed (p=0.052), Chao1 (p=0.082), Shannon (p=0.106) and Simpson (p=0.125)). Similarly, when we restricted our analysis to patients whose sputum communities had ≤25% Pseudomonas RA (n=66), patients who progressed to eESLD (8; 12%) had lower Observed (p=0.001) and Chao1 (p=0.002). The organism most strongly associated with protection against progression to eESLD in these subsets was Haemophilus. Indeed, Haemophilus was conspicuously absent as a microbiota constituent in patients progressing to eESLD when we assessed those patients who did not culture P. aeruginosa (RA eESLD: 0.009% (IQR 0.005–0.013) vs nESLD: 9.81% (IQR 0.81–47.2), p<0.001) and in those patients whose microbiota contained <25% Pseudomonas (RA eESLD: 0.009% (IQR 0.004–0.7) vs nESLD: 5.47% (IQR 0.51–28.7), p<0.001).
We identified six significant core genera with different RAs between eESLD and nESLD (table 2). In those with eESLD Pseudomonas RA was significantly greater, whereas Streptococcus, Haemophilus, Granulicatella, Gemella and Rothia were lower. Similar results were obtained when alternate definitions of progression to eESLD were used (prior to 23 or 27 years, DNS).
Relative abundance of classical CF pathogens and core CF microbiome constituents, as a function of clinical outcomes
Microbiota associated with secondary outcomes of interest: accelerated lung function decline
There was a trend towards reduced sputum alpha diversity in Ab-RP though it did not meet significance (figure 2B). When we did a sensitivity analysis using alternate definitions of decline, −2%/year, −2.5%/year and −3.5%/year, the same trends were evident (DNS). Similar results using a 2.5-year period were observed (DNS). When the samples were analysed by relative lung function decline, there were significant differences between Re-RP and Re-Nrp for all alpha-diversity estimators except Simpson index (figure 2C). However, no clustering of patients by microbiome was observed by rates of either absolute (Ab-RP vs Ab-Nrp; p=0.328) or relative decline (Re-RP vs Re-Nrp; p=0.07, PERMANOVA). Similar results were obtained when alternate definitions of Ab-RP were used, including: −2%/year, −2.5%/year and −3.5%/year (DNS).
With respect to individual microbial community constituents, the RAs of Granulicatella and Gemella were significantly greater (p<0.05) in Nrp compared with RP, for both absolute and relative decline (table 2). In addition, Rothia and Veillonella (p<0.05) were enriched in samples from Re-Nrp compared with Re-RP (table 2). Finally, Re-RP patients had higher RA of Fusobacterium as compared with the Re-Nrp patients (p=0.03) (table 2).
Exploring clinically translatable biomarkers to predict eESLD and accelerated lung function decline
To identify if specific microbiota profiles could be adapted as biomarkers for predicting future clinical disease progression, we examined associations within the microbiota (ie, alpha-diversity estimators, organism predominance) and disease progression groups. We found an association between alpha-diversity estimators with progression to eESLD or being an RP based on relative decline and the same trends (although not significant) for absolute (DNS). Because both Observed and Chao1 only estimate species richness, we decided to only further analyse the association of Simpson’s diversity index (SDI) and Simpson index, which take into account both species richness and evenness, with disease progression. Patients with SDI <1 or Simpson index <0.5 disproportionally progressed to eESLD (figure 4A). Similar results were found for the risk of being an Re-RP (figure 4B) but not Ab-RP (figure 4C). We developed a multivariable logistic regression model to predict progression to eESLD based on the selection of variables that were associated with the primary outcome. Factors independently associated with eESLD were baseline FEV1 and alpha diversity (table 3).
Logistic regression model predicting progression to eESLD in 18–22 year-old patients with CF

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Shannon and Simpson indexes as biomarkers to predict disease progression. Comparison of the proportion of patients with Shannon index >1 and Simpson index >0.5 in patients who progressed to early end-stage lung disease (eESLD) or not (nESLD) (A), in rapid progressors (RP) and non-rapid progressors (Nrp) patients based on the relative (B) and absolute (C) rates of lung function decline. Fisher’s exact test was conducted at two tailed. Ab, absolute; Re, relative; RR, risk ratio.
As Pseudomonas, Streptococcus, Staphylococcus and Haemophilus accounted for >80% of total reads, we compared these organisms at varying percentage of RA with disease progression groups. We found that patients with RA of Pseudomonas ≥50% or ≥75% had 2.8 and 4.88 times the risk to progress to eESLD (p=0.02 and p<0.001, respectively) compared with those nESLD patients (table 4). In contrast, patients with RA of Streptococcus ≥25% are associated with lower risk of eESLD and have a 32% and 41% reduction in risk of accelerated annual FEV1% decline measured relatively and absolutely, respectively. Next, we determined the association of core CF microbiome genera with clinical outcomes at a specific RA based on their frequency distribution (table 2). We found that higher levels of Granulicatella and Gemella are associated with lower risk of progression to eESLD and respiratory disease progression, measured by both absolute and relative measures of lung function decline (table 4).
Relation between the clinical outcomes and the specified percentage of relative abundance in some CF microbiota members
Next, we sought to determine if specific combinations of organisms combined with either Pseudomonas or Streptococcus RA associated with increased risk of progression to eESLD (table 4). Such an association with patient outcomes might suggest bacterial synergy or antagonism within CF airways. Several microbial combinations were statistically significant in terms of their association with the progression with eESLD (online supplementary table E1). We observed two general trends. Patients with RA of Pseudomonas ≥75% regardless of other community members were associated with higher risk to progress to eESLD. No additional organisms worsened clinical course. In contrast, sputum from patients with abundant Streptococcus in combination with other microflora further associated with lower risk to progress to eESLD (online supplementary table E1).
These same combinations were tested for the secondary outcomes (DNS) and it was observed that only patients with Granulicatella ≥0.75% or Gemella ≥0.75% combined with Streptococcus ≥25% had lower risk to be an RP compared with the Nrp for both absolute (risk ratio (RR): 0.6, p=0.0105 and RR: 0.58, p=0.006, respectively) and relative (RR: 0.37, p=0.0002 and RR: 0.45, p=0.002, respectively) lung function decline. Similarly, patients with Granulicatella ≥0.75% or Gemella ≥0.75% combined with Pseudomonas <25% are associated with lower risk of being an Re-RP (p=0.0034 and p=0.001, respectively).
Discussion
Identifying biomarkers to better delineate future clinical course has been a primary focus of CF research.20 Whereas efforts to explore the inflammasome have flourished, efforts to harness microbial biomarkers predicting lung disease have slowed. This is somewhat understandable given the marked heterogeneity in clinical course experienced by individuals infected with classical CF pathogens. Indeed, recent studies have even failed to demonstrate different outcomes for those with incident P. aeruginosa infections that progress to chronicity relative to those who are eradicated.2–4 Additionally, to establish certain organisms such as methicillin-resistant Staphylococcus aureus as a CF pathogen, data sets involving thousands of patients were required to demonstrate significant differences in FEV1 decline as smaller data sets did not.21 Furthermore, recent studies have failed to demonstrate correlation of classical microbiologic endpoints with subsequent outcomes.22 23 Thus, the recognition of a diverse and complex community inhabiting the lower airways proved enticing.24 Cross-sectional cohort studies have demonstrated patients with advanced lung disease are more likely to have microbial communities with limited diversity, dominated by classical CF pathogens.25 However, predictive studies are required to understand if the microbiome may be harnessed to predict clinical outcomes.
Our data demonstrate that young adults with sputum of lower alpha diversity have greater risk of progression to eESLD. Specifically, patients with SDI <1 or Simpson index <0.5 have the highest risk. Although both indices measure the richness and evenness (abundance and distribution of species among a community) there are some differences between metrics. In particular, the Simpson index has greater weighting on dominant species compared with SDI.26 Not surprisingly, we identified that proportional abundance of Pseudomonas within a community was a large driver of this effect, with patients who ultimately progressed to eESLD having both a higher RA and greater likelihood of having a population abundance ≥50% or ≥75%. The only other genus which trended towards a negative association with patient outcomes was Stenotrophomonas.27 We did not observe any organism in combination with Pseudomonas which appeared to increase the risk of progression to eESLD suggesting that the primary pathogen in CF lung disease remains P. aeruginosa—when abundantly present. However, this analysis may be insensitive to interspecies interactions known to occur between P. aeruginosa and microbiome constituents.28 29 Neither the presence of Pseudomonas in microbial communities nor the dichotomous binning of patients into P. aeruginosa culture positive/not (even when assessing mucoidy) was sensitive to establishing risk of progression to eESLD. Other traditionally accepted factors associated with progression to death/transplantation in CF were observed herein including baseline lung function and nutrition,30 31 but we were underpowered to detect others including CF-related diabetes and gender. Based on the multivariate model, we found that only alpha diversity as measured by SDI and baseline lung functions were independently associated with progression to eESLD.
We also assessed the microbiome in the context of subsequent pulmonary function decline. We assessed for both an absolute and relative rate of lung function decline to ensure sensitivity to detect changes in those with advanced disease.32 We chose a rate of >−3%/year as being disproportionate with expected and defined them as rapid decliners.33 34 We observed low microbial diversity as well as conventional factors associated with relative RP status including worse baseline lung function and body mass index, but we did not note observed changes associated with absolute RP status—although similar trends were evident. This is likely because those individuals with advanced lung disease did not have capacity to lose lung function >−3%/year. Zhao et al previously retrospectively assessed biobanked sputum from six CF men and observed that those patients with reduced microbial diversity were more likely to experience disease progression.6
The approach used in this study allowed us to assess microbial community of patients who progressed to eESLD or became RP. Several of the genera that associated with progression groups (ie, Granulicatella and Gemella) were found in lower RA compared with the top four genera (ie, Pseudomonas, Streptococcus, Haemophilus and Staphylococcus). Very low abundance OTUs may have important effects on the bacterial community structure and pathogenesis as has been observed in non-CF settings.35 Both Granulicatella spp and Gemella spp are facultative anaerobic species that have been associated with CF PEx.36 37 Interestingly, it has been suggested that Gemella spp may play a role in remodelling a normal CF microbiota into a dysbiotic one,24 as its RA increased during PEx in one study.38 Another study also showed a positive correlation between the RA of Granulicatella and higher FEV1 values during early treatments for PEx.39
We also identified that the presence of Streptococcus at an RA ≥25% reduced risk of rapid pulmonary function decline and end-stage lung disease. Few studies have focused on the role of Streptococcus spp on CF clinical outcome.40 However, recently a mechanism by which an oral commensal, Streptococcus parasanguinis, may interfere with the pathogenesis of P. aeruginosa was described.41 We did not observe Burkholderia to be significant factor in our cohort. However, of five patients culturing Bcc, four were Burkholderia multivorans and only one was Burkholderia cenocepacia (a non-epidemic strain that was subsequently eradicated) perhaps explaining the muted observed responses.42
While we described possible novel CF microbiome biomarkers for prediction of the disease progression herein, it is important to highlight that CF is a complex disease that occurs as a combination of microbial colonisation, host immune response, nutritional status and CF-related comorbidities, all of which are further confounded by patient factors including treatments, compliance, environment and socioeconomic status.43 The microbiome-based biomarkers described in this study better predicted progression to eESLD rather than the secondary outcome, accelerated lung function decline, highlighting the complexity of using lung function decline as it may be affected by extrinsic factors.
We acknowledge a number of limitations. This work represents retrospective analysis of prospectively collected sputum from 104 individuals from a single CF centre—whereas most pathogen epidemiological studies have required thousands of patients to demonstrate pathogenic potential.44 However, this is the largest targeted study of the CF microbiome—focusing specifically on individuals at a time of greatest risk of disease progression (young adulthood).9 10 Indeed, transition, when an entirely new care team is involved, may be an ideal time for a new technology to be trialled. Whether these results can be extrapolated to other age ranges is unknown. While our exclusion of samples from patients experiencing PEx/antibiotic therapies resulted in a modest reduction in cohort size, we have eliminated the confounding selective pressures of systemic antibacterials.45 While we focused our results on a single sample from each individual, we have validated that an individual’s microbiome is relatively consistent.11 While we have attempted to create a large homogeneous group by including 104 individuals of similar age, we included individuals from a period spanning almost 20 years—a potential concern recognising earlier cohorts decline at a greater rate.46 However, prior work from our group has not demonstrated major differences in the microbiome of young adults spanning these periods and neither our primary nor secondary outcomes were disproportionally observed in older samples.11 While our use of V3-V4 regions to identify microbiome constituents has only modest ability to identify OTU to the species, P. aeruginosa predominates. Indeed, of the 104 patients in our cohort none had ever a cultured non-aeruginosa Pseudomonas.
Conclusion
We observed that CF young adults with low microbiome diversity, dominated by the archetypal CF pathogen P. aeruginosa, are more likely to proceed to eESLD and/or subsequently experience exaggerated lung function decline. Conversely, those with a diverse community, dominated by organisms such as Streptococcus, were more likely to have a milder clinical course. These novel biomarkers could be used to identify patients with CF at increased risk of disease progression for more intensive monitoring and treatment. These results support large-scale collaborative multicentre studies evaluating the microbiome as a novel biomarker.
Acknowledgments
We thank the staff of the Calgary Adult CF Clinic and Calgary Laboratory Services for their continued efforts to stock and maintain the CACFC Biobank.
References
Footnotes
Contributors NA was primarily responsible for sputum sample collection, extraction and microbiome analysis. Sample analysis and statistical analyses were performed by NA, AH, MGS, MLW, CDS and RS. NA, HRR, CDS and MDP were responsible for collection and maintenance of the CACFC Biobank and clinical care records, and documentation. NA was responsible for the creation of the initial draft of the manuscript. All authors contributed to development of the final manuscript. MDP is the guarantor of this work.
Funding This study was supported by a grant from the Canadian Institute of Health Research to MDP, grant number 364568.
Competing interests MDP, HRR and MGS have received research funding from Gilead Sciences. MDP and HRR have participated in advisory boards and/or performed consulting work for Vertex but both declined honoraria. NA, AH, RS, MLW and CDS have no conflicts to declare.
Patient consent Not required.
Ethics approval Conjoint Health Research Ethics Board of the University of Calgary (REB-15-2744).
Provenance and peer review Not commissioned; externally peer reviewed.