Article Text

Abstract
Background The use of electronic (e)-cigarettes is increasing rapidly, but their lung health effects are not established. Clinical studies examining the potential long-term impact of e-cigarette use on lung health will take decades. To address this gap in knowledge, this study investigated the effects of exposure to aerosolised nicotine-free and nicotine-containing e-cigarette fluid on mouse lungs and normal human airway epithelial cells.
Methods Mice were exposed to aerosolised phosphate-buffered saline, nicotine-free or nicotine-containing e-cigarette solution, 1-hour daily for 4 months. Normal human bronchial epithelial (NHBE) cells cultured at an air-liquid interface were exposed to e-cigarette vapours or nicotine solutions using a Vitrocell smoke exposure robot.
Results Inhalation of nicotine-containing e-cigarettes increased airway hyper-reactivity, distal airspace enlargement, mucin production, cytokine and protease expression. Exposure to nicotine-free e-cigarettes did not affect these lung parameters. NHBE cells exposed to nicotine-containing e-cigarette vapour showed impaired ciliary beat frequency, airway surface liquid volume, cystic fibrosis transmembrane regulator and ATP-stimulated K+ ion conductance and decreased expression of FOXJ1 and KCNMA1. Exposure of NHBE cells to nicotine for 5 days increased interleukin (IL)-6 and IL-8 secretion.
Conclusions Exposure to inhaled nicotine-containing e-cigarette fluids triggered effects normally associated with the development of COPD including cytokine expression, airway hyper-reactivity and lung tissue destruction. These effects were nicotine-dependent both in the mouse lung and in human airway cells, suggesting that inhaled nicotine contributes to airway and lung disease in addition to its addictive properties. Thus, these findings highlight the potential dangers of nicotine inhalation during e-cigarette use.
- COPD ÀÜ Mechanisms
- Emphysema
- COPD Pathology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is the key question?
What are the pulmonary consequences of electronic (e)-cigarette inhalation?
What is the bottom line?
These findings show that the inhalation of nicotine present in e-cigarettes alters inflammation, ion conductance and mucociliary function in human bronchial epithelial cells and induces airway hyper-reactivity and air space enlargement in exposed mice.
Why read on?
This study provides insights into the lung health effects of e-cigarettes and also implicates nicotine as a causative factor at the onset and progression of COPD.
Introduction
Exposure to cigarette smoking is the leading cause of preventable death in the USA. The majority of COPD deaths are associated with smoke exposure, with COPD now ranking as the third leading cause of death in the USA1 and it is projected to become the third leading cause worldwide within the next 20 years.2 Quitting smoking lowers the risk for several diseases and can increase life expectancy.2 Though smoking prevalence in the USA has decreased over the past 50 years, approximately 19% of the population continues to smoke.3 Though nicotine replacement therapy (NRT) helps, nearly 93% of smokers will relapse despite NRT within 6 months of quitting.4 Given these difficulties with smoking cessation and the enormous public health burden imposed by COPD, some healthcare advocates have argued that electronic (e)-cigarettes are a safer alternative for those individuals who cannot quit.5
e-Cigarettes are devices that effectively deliver vaporised liquid nicotine to the lungs. The user can choose the nicotine concentration of e-cigarette liquid (e-liquid) that is loaded into the device's cartridge. When the user inhales, the e-liquid, primarily nicotine in propylene glycol (PG) or vegetable glycerine (VG), is heated to produce a vapour that is inhaled into the lungs. Despite the many flavours and varieties available, almost all consumers choose e-liquids that contain nicotine.6 Recent studies indicate that e-cigarette exposure could pose several future health problems. The use of e-cigarettes enhances oxidative stress and inflammation in mice7 and impairs immune defence against bacterial and viral infection.8 This also has implications in secondhand smoke exposure as e-cigarettes significantly increase the nicotine content of indoor air and expose non-users to secondhand vapours.9 Nicotine enhances airway hyper-reactivity in lipopolysaccharide-challenged mice and inflammation in lung epithelial cells.10 Nicotine also suppresses apoptosis in lung tumours.11 However, the biological effects of nicotine delivered to the lungs by e-cigarettes have not been thoroughly studied.
Here, we used in vitro and in vivo models to evaluate the biological impact of e-cigarette usage on mouse lungs and in normal human bronchial epithelial (NHBE) cells collected from a series of human subjects. This study found that inhaled nicotine in e-cigarettes triggered effects associated with the pathogenesis and progression of COPD: airspace enlargement, mucous cell hypertrophy, release of inflammatory mediators and altered epithelial ciliary function. The data collected provide direct evidence of the potential harmful effects of e-cigarettes and, in particular, nicotine inhalation into the lung.
Methods
In vivo inhaled exposure to e-cigarette fluids
All animal experiments were performed with approval from Mount Sinai Roosevelt Medical Center's Institutional Animal Care and Use Committee. We developed an in vivo e-cigarette exposure model using an mouse pie cage (MPC) Aerosol Medication Nebulizer (Braintree Scientific, Braintree, Massachusetts, USA). For 4 months (5 days/week), A/J mice aged 12 weeks (Jackson labs) were whole body exposed to 0.4 mL of phosphate-buffered saline (PBS) or e-cigarette vehicle (PG and VG 50/50; American eLiquid Store) containing 0 or 18 mg/mL nicotine (American eLiquid Store). All liquids were preheated to 37°C prior to nebulisation and the exposures lasted 1 hour. Mice were euthanised 24 hours after the last exposure. Bronchoalveolar lavage fluid (BALF) was collected and immune cells quantified as previously described.12 Plasma cotinine levels were measured using a commercially available kit and following the manufacturer's instructions (Abnova, Walnut, California, USA).
Airway responses to methacholine challenge
Airway responses to increasing doses of methacholine (Sigma Chemical, St. Louis, Missouri, USA) were assessed with the Scireq Flexivent system (Scireq, Montreal, Quebec, Canada) in mice exposed to 4 months of PBS and e-liquid containing 0 or 18 mg/mL nicotine. Animals were anaesthetised with ketamine/xylazine (10 mg/kg) and paralysis was induced with 1 mg/kg pancuronium bromide intraperitoneally. Baseline pulmonary function measurements (forced expiratory flow at 50% of FVC (FEF50)) were determined prior to methacholine challenge in mice. The linear single-compartment model was used to assess total respiratory system resistance.
Histological analysis
The lungs underwent pressure-fixation and fixed tissue was processed and H&E stained for analysis of morphometry as previously described.13 Mucin was stained with Alcian blue and counterstained with Safranin O using a commercially available kit (Abcam, Cambridge, Massachusetts, USA). For immunofluorescence, slides were blocked with 10% goat serum in PBS for 1 hour at room temperature. The slides were incubated with primary antibody (rabbit polyclonal anti-MUC5AC IgG, 1:50 dilution; Santa Cruz Bio) or rabbit IgG negative control in 5% goat serum 1% bovine serum albumin in PBS overnight at 4°C and then with secondary antibody (donkey antirabbit IgG-546, 1:200 dilution) for 1 hour at room temperature. Immunoreactivity was visualised with an epifluorescence microscope. Tissue quantification of macrophages, neutrophils and lymphocytes were detected on slides with primary antibodies; rabbit polyclonal anti-MAC3, antineutrophil elastase or anti-CD3 IgG (Abcam; 1:50 dilution). Immune cells were quantified from 10 randomly selected ×400 images of the lung tissue sections from each mouse.
Cell viability analysis
Apoptosis was determined on paraffin-embedded tissue by the terminal deoxynucleotidyl transferase dUTP nick end-labelling (TUNEL) in situ cell death detection kit AP (Roche Diagnostics), following the manufacturer's instructions. For each lung, 10 random sections were obtained at three different depths of tissue sectioning and a minimum of 1000 cells/section were visually evaluated after staining. The positively labelled cells were expressed as a percentage of total nuclei. Caspase 3/7 activity was measured as previously described,14 using a commercially available Caspase-Glo 3/7 Assay System (Promega, Fitchburg, Michigan, USA). In vitro cell viability was determined by lactate dehydrogenase (LDH) released into media using a commercially available assay (Sigma Aldrich).
qPCR, MMP and cytokine protein detection
RNA was isolated using the Qiagen RNeasy kit and complementary DNA (cDNA) was reverse transcribed using the Applied Biosystems high-capacity cDNA kit. qPCR was performed on the Bio-Rad CFX384 real-time system using validated TaqMan probes (Life Technologies/Applied Biosystems). qPCR results are represented as relative quantification. Gene expression was normalised to β-actin or glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Cytokine protein levels were measured in culture media using a beads assay on a Bio-Rad Bio-Plex 200 system (Bio-Rad, Hercules, California, USA). Collagenase units were calculated as the number of µmoles of leucine liberated per mg of enzyme, by a colorimetric ninhydrin method.15 Cathepsin activity assays were performed as previously described.16
Immunoblot analysis
Lung tissue was homogenised in four volumes of radioimmunoprecipitation assay buffer. Equal concentrations of protein were immunoblotted for phosphorylated PKCα (p-PKCα), PKCα, phosphorylated extracellular regulated kinase (ERK), ERK and β-actin (Cell Signaling Technology, Danvers, Massachusetts, USA). Chemiluminescence was detected and quantified with a Molecular Imager ChemiDoc XRS+ (Bio-Rad). Densitometry was performed and represented as a ratio of pixel intensity of the phosphorylated protein compared with total protein, using Bio-Rad Laboratories Image Lab software (V.4.0, build 16).
In vitro analyses of NHBE cells
NHBE cells were isolated from appropriately consented donors whose lungs were found unsuitable for transplantation. The 15 donors were non-smokers with no known airway diseases, 5 females and 10 males aged 25–58 years. From a racial aspect, there were 10 Caucasians, 2 African-Americans, 1 Hispanic white and 2 unknowns. After expansion, cells were redifferentiated at the air–liquid interface (ALI) on collagen-coated 12-mm Transwell inserts (Corning, Lowell, Massachusetts, USA).17–19 The cells were exposed to 36 puffs of vaporised e-liquid either containing 0 or 36 mg/mL nicotine using the VC-1 exposure system (Vitrocell Systems, Waldkirch, Germany), with a 70 mL puff volume applied every 30 s. Cells were also exposed to nicotine (0 or 100 μM, with an estimated final 100 nM nicotine concentration in the airway surface liquid (ASL) after nebulisation) aerosolised in the Cloud system (Vitrocell Systems) or baseolaterally supplemented nicotine (0, 1 or 10 μM) for 12 hours or 5 days.
Ion channel function and mucociliary clearance
Short-circuit currents (Isc) and ciliary beat frequency (CBF) were recorded at the following time points: 8 hours after vapour exposure in the VC-1 system, 4 hours after nebulised nicotine in the Cloud system and 12 hours after basolateral addition of nicotine.20 Cystic fibrosis transmembrane regulator (CFTR) function was determined by the decrease in Isc after addition of CFTR-inh in cells stimulated with 10 µM forskolin in the presence of 10 µM amiloride at room temperature. Apical, ATP-stimulated K+ (BK) currents were measured as previously described.20
Measurement of ASL and percent ciliated surface
Twenty-four hours after exposure of cells on 12 mm inserts to 36 puffs of e-cigarette vapour with 0 or 36 mg/mL nicotine in the VC-1 exposure system, the cultures were imaged with an Epson flatbed scanner and images were analysed for meniscus volume using ImageJ software (National Institutes of Health, Bethesda, Maryland, USA), kindly provided by Michael Myerburg (University of Pittsburgh). Fully redifferentiated NHBE cells were fixed, blocked and stained with a monoclonal antitubulin acetylated antibody (1:10 000 dilution; Sigma Aldrich) to detect cilia. The % ciliated area was determined using ImageJ software.
Statistical analyses
F-tests, D'Agostino-Pearson Omnibus normality tests, non-parametric Friedman test and Student's t-test were performed on all data sets. One-way analysis of variance was used for comparisons of more than two groups followed by a post hoc test for linear trend analysis on human cell samples. Data are represented as dot plots with a line denoting the mean and bars denoting the SEM. GraphPad Prism Software (GraphPad, La Jolla, California, USA) was used for all data analysis and graphical representations.
Results
Exposure to inhaled nicotine-containing e-cigarette fluids induces airway hyper-reactivity and airspace enlargement
Mice were exposed to aerosolised PBS and e-liquid (0 and 18 mg/mL nicotine) daily for 4 months. Exposure to aerosolised nicotine-containing e-cigarettes increased plasma cotinine, the nicotine degradation product, demonstrating that nicotine was effectively delivered to the lungs and absorbed systemically by the mice (figure 1A). Airway hyper-reactivity was assessed by methacholine challenge test. At every methacholine dose ≥1 mg/mL, mice exposed to nicotine-containing e-cigarette fluids showed significantly higher respiratory resistance than mice exposed to aerosolised PBS or to e-cigarette fluids that did not contain nicotine (figure 1B and online supplementary figure S1). No differences in airway hyper-reactivity were found in mice exposed to nicotine-free e-cigarette fluids when compared with mice exposed to PBS. To examine baseline lung function changes due to e-cigarette use, forced expiratory flow at 50% (FEF50) of FVC was determined. The FEF50/FVC ratio was significantly reduced from 23±1.2 in mice exposed to nicotine-free e-cigarette fluids to 15±1.5 in mice exposed to e-cigarettes containing 18 mg/mL nicotine (average±SEM; p<0.01), suggesting obstruction of the small airways (figure 1C).

Lung function in mice exposed to inhaled phosphate-buffered saline (PBS), nicotine-free and nicotine-containing e-cigarette fluids. Mice were exposed daily to inhaled PBS, nicotine-free (EC vehicle, 0 mg/mL) or nicotine-containing (EC vehicle, 18 mg/mL) e-cigarette fluid for 4 months and nicotine absorption was confirmed by (A) plasma cotinine levels. (B) Airway hyperresponsiveness to increasing doses of methacholine was assessed in each animal group. (C) Negative pressure-driven forced expiratory and forced oscillation technique manoeuvres were performed to determine changes in pulmonary mechanics in all animal groups. Forced expiratory flow at 50% of FVC (FEF50) were determined in each animal. (D) Representative H&E-stained images of fixed lung parenchyma from mice exposed daily for 4 months (scale bar=100 µm) and quantification of surface area/unit volume, fractional volume and mean linear intercept. Graphs are represented as mean±SEM of three measurements. p Values shown, comparing both treatments connected by a line. All n≥6 per group. The comparisons between two individual groups were determined by Student's t-test. EC, e-cigarette.
Supplementary figures
Mice exposed to inhalation of nicotine-containing e-cigarette fluids showed significant airspace enlargement, while nicotine-free e-cigarette fluids or PBS exposure had no impact on lung architecture (figure 1D). Mean linear intercept, surface area/unit volume ratio and fractional volume were calculated in H&E-stained lung sections. All three parameters were altered in the mice exposed to nicotine-containing e-cigarette fluids: mean linear intercept was increased and surface area/unit volume and fractional volume were decreased (p<0.05 for all; figure 1D). No difference in any of these parameters was observed between mice exposed to nicotine-free e-cigarette fluids or PBS (figure 1D).
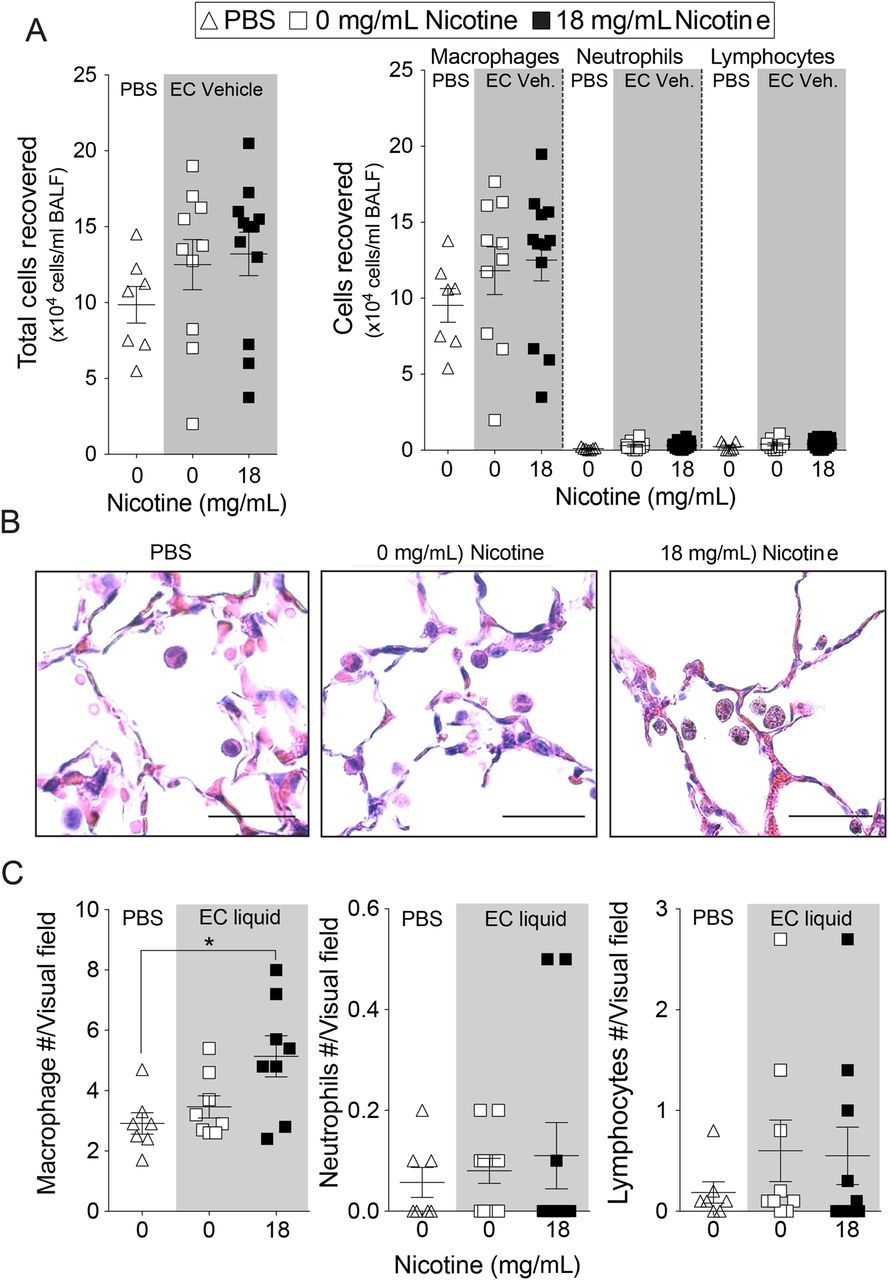
Nicotine inhalation enhanced inflammatory cell infiltration into the lung. After 2 weeks of exposure, BALF cells increased from 130 000±27 000 cells/mouse in the mice exposed to nicotine-free e-cigarette fluids to 280 000±41 000 cells/mouse in the mice exposed to nicotine-containing e-cigarette fluids (p<0.01; mean±SEM; online supplementary figure S2D). After 4 months of exposure, mice exposed to nicotine-containing e-cigarette fluids showed a trend towards increased cell number in BALF (figure 2A). Cytospins showed that macrophages were the most abundant cell type recovered in the bronchoalveolar lavage in all three conditions, while neutrophil and lymphocytes constituted a minor proportion (figure 2A). Macrophages were observed in the alveolar space in all mouse groups (figure 2B) and quantification of tissue inflammation demonstrated that elevated macrophage numbers in lung tissue of mice subjected to nicotine-containing e-cigarette fluids compared with the other groups (figure 2C). Neutrophil and lymphocyte numbers were not altered (figure 2C).
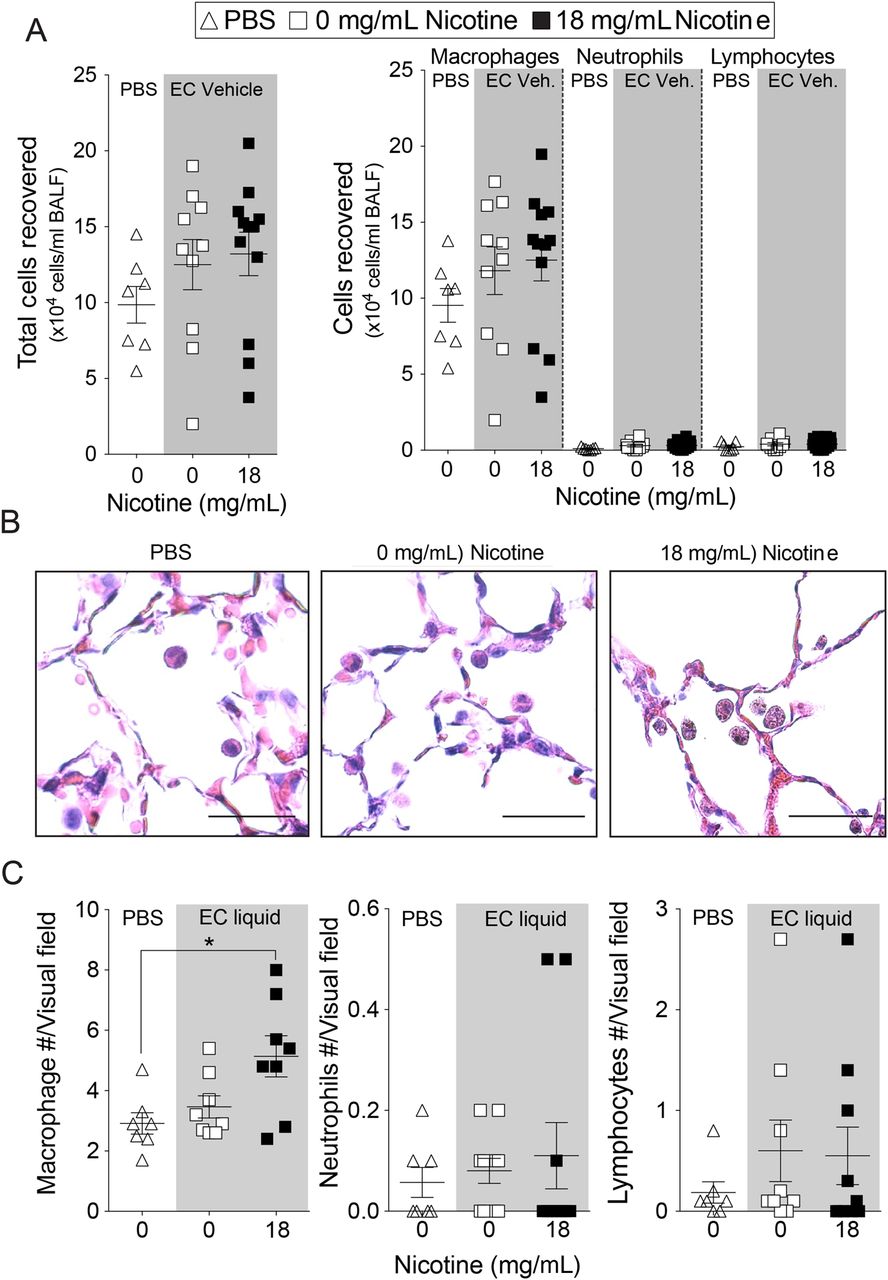
Lung inflammation in mice exposed to inhaled phosphate-buffered saline (PBS) and to nicotine-free e-cigarette fluids. Mice were exposed daily to inhaled PBS, nicotine-free (0 mg/mL) or nicotine-containing (18 mg/mL) e-cigarette fluids for 4 months. (A) Total bronchoalveolar lavage fluid cells, macrophages, neutrophils and lymphocytes were quantified in mice exposed daily for 4 months. (B) Representative H&E-stained images of fixed lung parenchyma and macrophages from mice exposed daily for 4 months (scale bar=20 µm). (C) Tissue cellularity was quantified by measuring the average number of macrophages, neutrophils or lymphocytes per high powered field (×400). Ten randomly selected high powered images were used for quantification per mouse. Graphs are represented as mean±SEM of three measurements. p Values shown, comparing both treatments connected by a line. All n≥7 per group. The comparisons between two individual groups were determined by Student's t-test. BALF, bronchoalveolar lavage fluid. EC, e-cigarette.
Nicotine in e-cigarette fluids triggers mucin production and apoptosis
Airway mucin production increased in mice exposed to nicotine-containing e-cigarette fluids, as shown by Alcian blue staining and fluorescence microscopy for MUC5AC (figure 3A). Mice exposed to nicotine-containing e-cigarette fluids showed a twofold increase in Muc5ac gene expression (p<0.01; figure 3A). Apoptosis of structural cells in the lung is another important event in the pathogenesis of COPD.21 Apoptosis, as estimated by TUNEL staining, increased within the alveoli and the airway epithelium of mice exposed to e-cigarette fluids that contained nicotine (figure 3B). Caspase 3/7 activity, increased from 37±1.5 units of luminescence in nicotine-free e-cigarette fluid exposed mice to 53±4.7 units of luminescence in nicotine-containing e-cigarette fluid exposed mice (p<0.01; mean±SEM; figure 3B). Taken together, nicotine in e-cigarettes promotes airway hyper-reactivity, inflammation, airway enlargements, mucus production and lung cell apoptosis.

Histological analysis of lungs from mice exposed to inhaled nicotine-free and nicotine-containing e-cigarette aerosol. Mice were exposed daily to inhaled nicotine-free (0 mg/mL) or nicotine-containing (18 mg/mL) e-cigarette aerosol for 4 months. Then, lungs were fixed and sectioned and stained for histological analysis. (A) Representative pictures of sections after Alcian blue stain and MUC5AC immunofluorescence (scale bar=20 µm). Graphs representing (A) qPCR analysis for Muc5ac are shown. (B) Representative images of sections after transferase dUTP nick end-labelling (TUNEL) (green) and DAPI (blue) staining from airway and alveolar regions of the lung (scale bar=20 µm). Percentage of TUNEL-positive cells quantified in each group and caspase 3/7 activity in lung tissue lysate are also shown. LU, luminescence units. Graphs are represented as mean±SEM of three measurements. p Values shown, comparing both treatments connected by a line. All n≥5 per group. The comparisons between two individual groups were determined by Student's t-test. DAPI, 4′,6-diamidino-2-phenylindole.
E-cigarette fluids containing nicotine induce lung cytokine and protease expression
Expression of several cytokines and proteases associated with tissue remodelling was measured in lung homogenates after 2 weeks (see online supplementary figure S2A–C) and 4 months of exposure (figure 4). Compared with exposure to inhaled e-cigarette fluids that lacked nicotine (0 mg/mL), e-cigarette fluid containing nicotine (18 mg/mL) caused a significant increase in the expression of Il-1β, monocyte chemoattractant protein-1 (MCP-1), Cxcl10, Il-6 and Cxcl2 (p<0.05 for all; figure 4A). Matrix metalloproteinase (MMP)-9 and MMP-12 and cathepsins K and L1 also showed increased messenger RNA expression in the mice exposed to e-cigarette fluids containing nicotine (figure 4B). Collagenase BALF activity increased by 70% (p<0.03) and cathepsin BALF activity showed a trend towards an increase in the BALF of mice exposed to e-cigarette fluids that contained nicotine (figure 4C). Expression of Mcp-1, Muc5ac and CatK showed similar patterns after 2 weeks of exposure to nicotine-containing e-cigarettes (see online supplementary figure S2). Our findings indicate that inhalation of nicotine via e-cigarette fluids can induce cytokine and protease expression and activity in a similar manner to tobacco cigarette smoke exposure.

Cytokine and protease expression and activity in lungs from mice exposed to nicotine-free or nicotine-containing e-cigarette fluids. Mice were exposed to inhaled nicotine-free (0 mg/mL) or to nicotine-containing (18 mg/mL) e-cigarette fluid for 4 months. Gene expression analysis by qPCR for (A) the cytokines IL-1β, MCP-1, CXCL10, IL-6, CXCL2 and CXCL5 and (B) proteases MMP-3, MMP-9, MMP-12, CatK, CatL1 and CatS. (C) Collagenase and cathepsin activity were measured in lung bronchoalveolar lavage fluid. Graphs are represented as mean±SEM of three measurements. p Values shown, comparing both treatments connected by a line. All n≥5 per group. The comparisons between two individual groups were determined by Student's t-test.
Inhalation of nicotine in e-cigarette fluids activates PKCα/ERK signalling in the lung
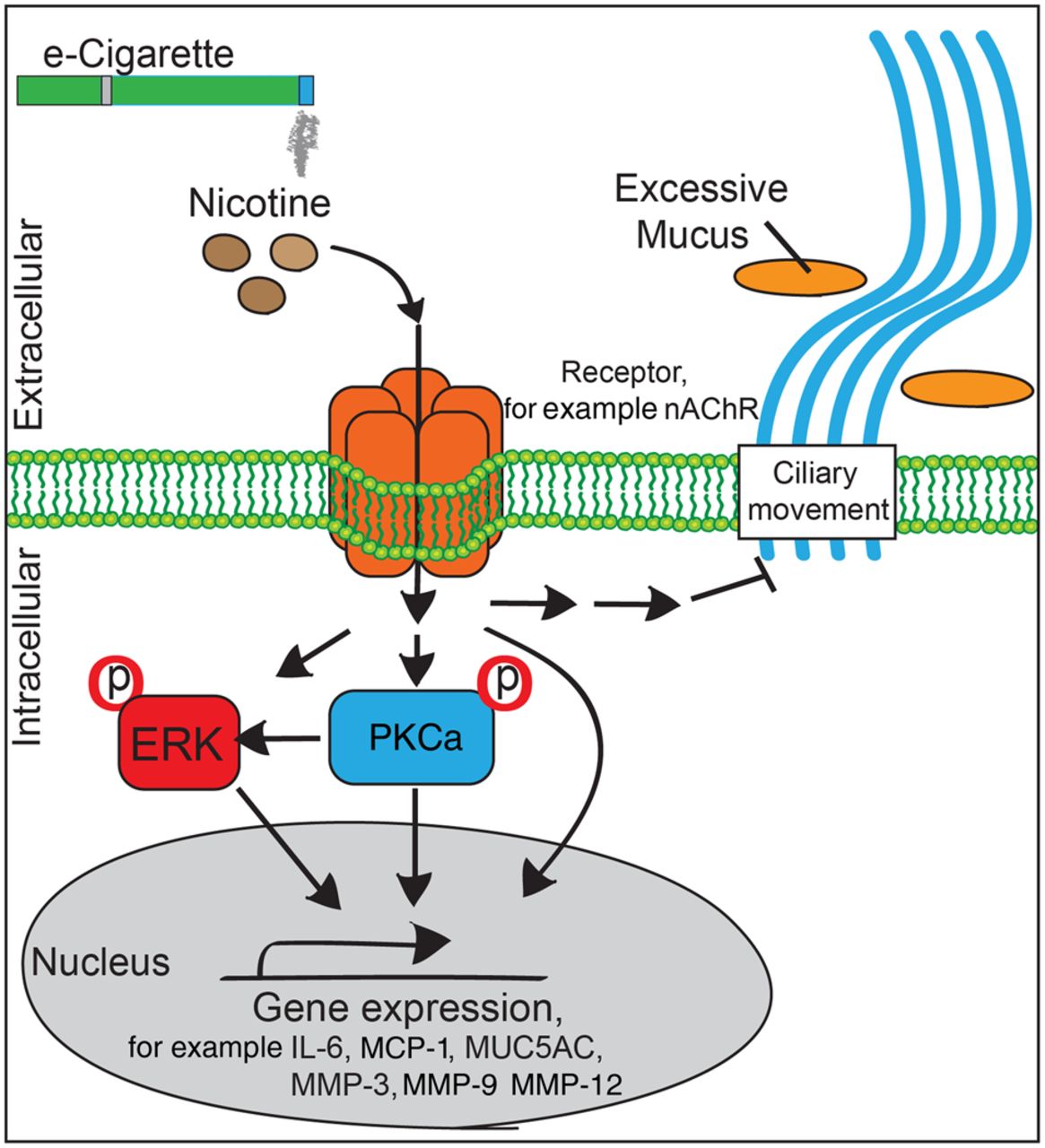
Our laboratory has previously shown that activation of PKCα and ERK by cigarette smoke regulated lung inflammation and protease responses.22 Since nicotine can stimulate the α-7-nicotinic receptor (α7nAchR) upstream of PKCα and ERK in human lung epithelial cells,11 ,23 we asked whether nicotine in e-cigarette fluids would activate this signalling mechanism. Lung homogenates from mice exposed to nicotine in e-cigarette fluids showed increased phosphorylation of PKCα and a trend increase in lung phosphorylation of ERK (figure 5). Of note, inhibition of this pathway prevents airspace enlargement in the lungs of cigarette smoke-exposed mice.22

PKCα and ERK activation in the lungs of mice exposed to inhaled vehicle or nicotine-containing e-cigarette fluid. Mice were exposed daily to inhaled nicotine-free (0 mg/mL) or to nicotine-containing (18 mg/mL) e-cigarette fluid for 4 months. (A) Representative western blots for total and phosphorylated PKCα (p-PKCα) and ERK in lung tissue homogenates. β-Actin was used as a loading control. (B) Optical intensity quantification and calculated ratios of phosphorylated/total protein for PKCα and ERK. Graphs are represented as mean±SEM of three measurements. p Values shown, comparing both treatments connected by a line. All n=9 per group for densitometry analysis. The comparisons between two individual groups were determined by Student's t-test.
The nicotine present in e-cigarette fluids alters ciliary function and cytokine production in NHBE cells
We used fully differentiated cultures of NHBE cells to assess the impact of nicotine-containing e-cigarette vapour on ciliary function. NHBE cells were obtained from healthy donors, cultured at the ALI and exposed to nicotine-free or to nicotine-containing e-cigarette vapours (figure 6A). Exposure to vaporised e-cigarette vapour had no effects on NHBE cell viability, determined by LDH assays (see online supplementary figure S3A). Ciliary beating serves as one of the main defence functions of the airways.24 Cells exposed to nicotine-containing e-cigarette vapours had significantly reduced CBF 8 hours after exposure (5.2±0.5 Hz vs 2.6±1 Hz; p=0.01; p<0.05; figure 6B). Since exposure to nicotine in e-cigarettes reduced CBF, the per cent of cilia on the apical cell surface was determined on NHBE cells. There was no significant difference in percentage of ciliary area on the apical surface 24 hours after exposure to nicotine-containing e-cigarette vapour (figure 6C). However, the gene expression of a major regulator of ciliogenesis, FOXJ1, was reduced in cells exposed to nicotine-containing e-cigarette vapours (figure 6D). Ion channel conductance for CFTR was significantly reduced in cells exposed to nicotine-containing e-cigarette vapours 8 hours after exposure (figure 6E), and was independent of the expression of the CFTR gene (figure 6F). Ion channel conductance for the BK channel was also significantly reduced in cells exposed to nicotine-containing e-cigarette vapours at 8 hours after exposure (figure 6G), which coincided with reduced expression of BK channel α subunit (KCNMA1) gene that regulates ASL.25 ASL volume changes showed a significant decrease, from 14.6±2 µL in cells exposed to nicotine-free e-cigarette vapour at 24 hours after exposure to 3.8±1 µL in cells exposed to nicotine-containing e-cigarette vapour (mean±SEM; p<0.01; figure 6I). Therefore, nicotine in e-cigarette vapour directly influences ciliary function.
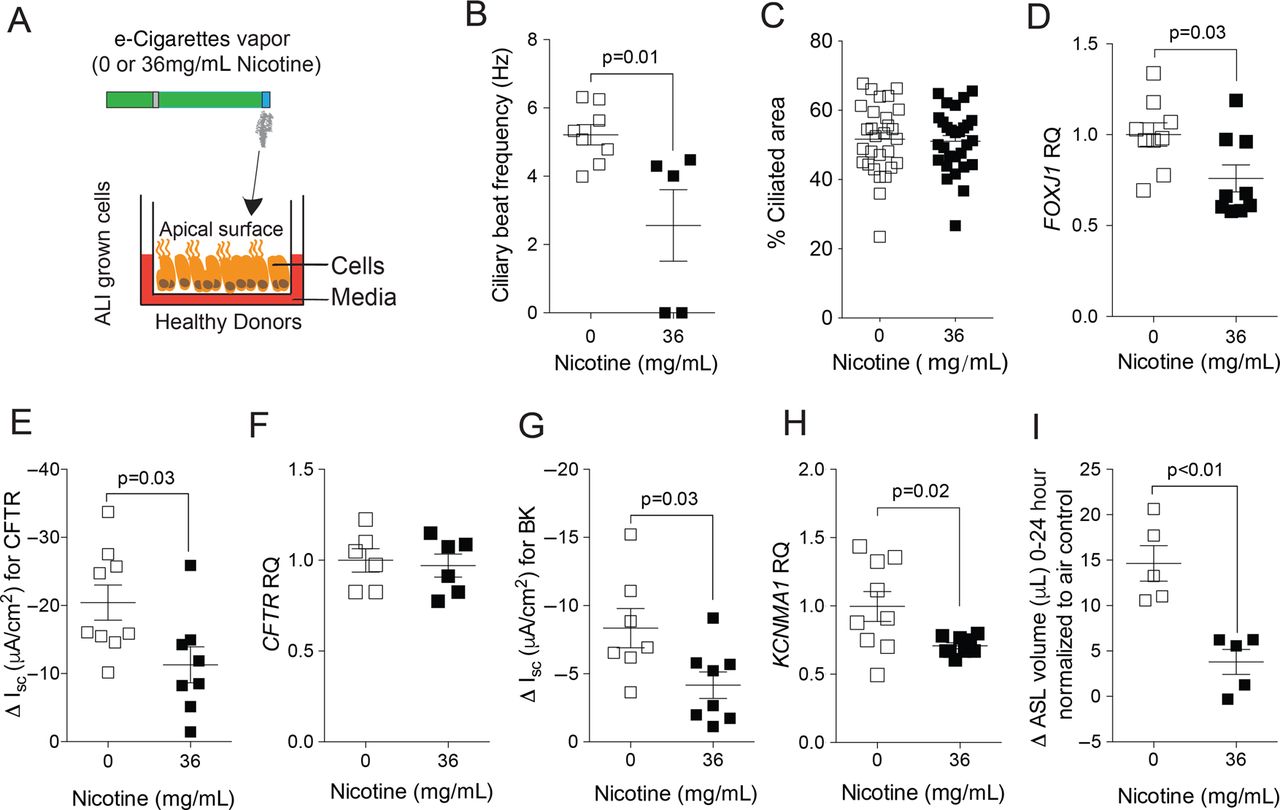
Effects of vehicle and nicotine-containing e-cigarette vapour on normal human bronchial epithelial (NHBE) cells. (A) Scheme showing how fully differentiated NHBE cells, grown at an air–liquid interface (ALI), were exposed to vapour from vehicle (0 mg/mL) or from nicotine-containing (36 mg/mL) e-cigarettes. (B) Ciliary beat frequency significantly decreased after exposure to nicotine-containing e-cigarette vapour. (C) Cells were stained with DAPI and antiacetylated tubulin to visualise cilia, 24 hours after exposures. The % ciliated area was determined using ImageJ software. (D) qPCR for gene expression of FOXJ1. (E) Cystic fibrosis transmembrane regulator (CFTR) ion conductance is significantly decreased after exposure to nicotine-containing e-cigarette vapour. (F) qPCR for gene expression of CFTR. (G) ATP-stimulated K+ (BK) ion conductance is significantly decreased after exposure to nicotine-containing e-cigarette vapour. (H) qPCR for gene expression of the α subunit of the BK channel (KCNMA1). (I) Measurement of airway surface liquid volume changes by refraction light microscopy. Graphs are represented as mean±SEM. p Values shown, comparing both treatments connected by a line. All n≥5 per group. The comparisons between two individual groups were determined by Student's t-test.
Nicotine directly alters ciliary function and cytokine production in NHBE cells
To examine how nicotine alone affected ion transport and CBF, NHBE cells were exposed to 0 or 100 µM nicotine nebulised (200 µL) into a chamber (Cloud from Vitrocell) (figure 7A). Using a Vitrocell microbalance, we determined that there was a total deposition of 25 nL in each well of our tissue culture plates. Assuming an ASL volume at baseline of about 40 µL, we calculated that the effective nicotine concentration would reach 60–120 nM in the apical surface liquid of these cells grown at ALI. Exposure to aerosolised nicotine had no effects on NHBE cell viability at 4 hours after nebulisation (see online supplementary figure S3B). Cells exposed to aerosolised nicotine showed decreased CFTR and BK conductance at 4 hours after exposure (figure 7B, C).

Effects of aerosolised nicotine on normal human bronchial epithelial (NHBE) cells. (A) Scheme showing how fully differentiated NHBE cells, grown at an air–liquid interface (ALI), were exposed to aerosolised saline containing 0 or 100 μM nicotine. Final concentration of nicotine in the airway surface liquid was estimated between 60 and 120 nM by microbalance deposition measurements. (B) Cystic fibrosis transmembrane regulator (CFTR) and (C) ATP-stimulated K+ (BK) ion conductances are significantly decreased in nicotine-exposed cell cultures compared with nicotine-free saline. Graphs are represented as mean±SEM. p Values shown, comparing both treatments connected by a line. All n≥4 per group. The comparisons between two individual groups were determined by Student's t-test.
To analyse the effects of long-term exposure to nicotine, NHBE cells were exposed to basolateral media supplemented with 0, 1 or 10 µM nicotine for 5 days (figure 8A). Exposure to nicotine in the culture media for 5 days had no effects on NHBE cell viability (see online supplementary figure S3C). In cells exposed to 10 µM nicotine in the culture media, CBF decreased from 7.2±0.3 in controls to 6.2±0.3 (mean±SEM; p<0.05; figure 8B) and there was a decreasing trend in the CFTR activity (figure 8C). Media nicotine supplementation also increased cytokine release both to the basolateral and the apical sides of the NHBE cell cultures at concentrations of 10 µM: interleukin (IL)-6 concentration in the wash increased from 390 to 442 pg/mL (p<0.05; median). IL-8 and MCP-1 also showed a trend towards an increase in the apical wash (figure 8D). In the media, IL-6 and IL-8 were significantly elevated by 10 µM nicotine exposures and MCP-1 trended towards an increase in nicotine-exposed cells (figure 8E). Together, these data suggest that nicotine exposure directly impacts parameters of mucociliary function and promotes a pro-inflammatory state in NHBE cells.

Cystic fibrosis transmembrane regulator (CFTR) conductance, ciliary beat frequency (CBF) and cytokine expression in normal human bronchial epithelial (NHBE) cells exposed to basolateral nicotine. (A) Differentiated NHBE cells, grown at an air–liquid interface (ALI), were exposed to nicotine (0, 1 and 10 μM) basolaterally. (B) CBF and (C) CFTR ion conductance measurements were performed 12 hours after exposure. Graphs are represented as mean±SEM. One-way analysis of variance was used to compare the time-course curves and multiple comparisons were determined by the Dunn's method. (D–E) NHBE cells were exposed to culture media with or without nicotine for 5 days as outlined in Methods section. The concentration of interleukin (IL)-6, IL-8 and MCP-1 was measured in the apical wash (D) and in the basolateral media (E) following 5 days of exposure to culture media with or without nicotine. Graphs are represented as median±range. p Values shown, comparing both treatments connected by a line; n≥3 per group. (D–E) Data sets were analysed by non-parametric Friedman tests.
Discussion
The Royal College of Physicians in the UK has recently advocated that switching to e-cigarettes will aid smoking cessation and reduce harm from conventional cigarette use.26 Indeed, it is well established that e-cigarettes produce less environmental pollutants, lack harmful tar and contain far fewer cancer-causing chemicals.9 ,27 Public health experts have argued that e-cigarettes will save lives by reducing or eliminating exposure to harmful cigarette components. This position, however, remains controversial as scientific data about the effects of e-cigarettes are lacking. Indeed, the Food and Drug Administration in the USA has announced that it will begin regulating e-cigarette products. This study shows for the first time that e-cigarettes induce pathogenic responses akin to what occurs in human COPD. Even more importantly, it demonstrates that nicotine is the key component responsible for these pathogenic changes in the lung. While e-cigarette usage may potentially cause less cancer and vascular disease compared with conventional cigarettes, our data indicate that it will still promote the development of obstructive airways disease. Thus, the findings from this study should be taken into consideration when advising patients on possible health risks of these products.
Traditionally, nicotine has been studied as the addictive component of cigarettes, but increasing evidence indicates that nicotine is also a key factor in the initiation and progression of COPD. Nicotine hinders the hydration of mucus,28 promotes Ca2+ influx in airway smooth muscle cells,29 blocks neutrophil apoptosis30 and induces pro-inflammatory dendritic cell responses.31 Smoke generated from high-dose nicotine cigarettes induces more emphysematous changes than low-dose nicotine cigarettes, in elastase-treated rats.32 The lack of focus on nicotine in this disease is surprising since genetic analyses identified nicotinic receptors (α3nAchR and α5nAchR) as susceptibility loci for COPD.33 Moreover, nicotine makes up almost 3% of the dry weight of a tobacco leaf and smokers breathe in 42 mg nicotine/day.34 Once absorbed, nicotine binds to nicotinic acetylcholine receptor (nAchRs). The airway epithelium expresses α3, α4, α5, α7, α9, β2 and β4 subunits for nAchRs,35 and their expression are highest on the apical cell surface, where exposure to inhaled nicotine occurs.36 In bronchiole cells, nicotine acted via α7AchR to upregulate γ-aminobutyric acid (GABA)ARα2 and induces Muc5ac expression and mucus production.37 This effect would explain the increased mucin staining and Muc5ac expression detected in mouse lungs following exposure to nicotine-containing e-cigarettes. Interestingly, CFTR and BK ion conductance and CBF were more negatively affected at a greater magnitude in cells exposed to aerosolised nicotine compared with cells where nicotine was supplemented in media. This indicates that apical exposure could be more detrimental than basolateral exposure. This occurred despite the fact that the concentration of nicotine was actually higher in the media (10 μM) compared with the ASL (60–120 nM) of the aerosol-exposed cells. This would suggest that inhaled nicotine impacts on the lungs differently than nicotine circulating in the body. However, further studies are needed to compare the pulmonary consequences of inhaled nicotine versus nicotine absorbed systemically though products like nicotine patches. Though nicotine acts via α7nAchR to stimulate PKCα (see figure 9 for potential signalling), it remains to be determined how much PKCα activation contributed to the pathogenic changes that occurred in our lung models. Indeed, identifying the signalling mechanisms responsible for these nicotine-mediated effects will be a key goal of future studies.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Possible signalling mechanism for nicotine-containing e-cigarette-induced lung damage. ERK, extracellular regulated kinase; IL, interleukin; nAChR, nicotinic acetylcholine receptor.
Other in vitro and in vivo studies that showed that short-term e-cigarette vapour inhalation alters lung inflammatory responses.7 ,8 To date, no other studies have looked at the effects of chronic e-cigarette exposure on lung inflammation in mice. Unlike our study and the study by Lerner et al,7 Sussan et al8 did not identify increases in IL-6 or MCP-1 in response to e-cigarette exposure but this may be accounted for by differences in mouse background, exposure time, e-cigarette flavouring, cotinine levels in plasma and the presence of a pulmonary infection.8 In ova-albumin sensitised mice, a 10-week e-cigarette exposure was reported to increase airway hyper-reactivity38 and we found that chronic e-cigarette inhalation, by itself, induces airway hyper-reactivity. Similar to cigarette smoke, nicotine in e-cigarettes promotes airway smooth muscle proliferation and contraction, which can impact airway hyper-reactivity in exposed individuals.39 Thus, nicotine could be a key factor driving the onset and progression of lung disease.
Commercially available, nicotine-free, vehicle was used as a control for the nicotine-containing e-cigarette liquid. With the exception of nicotine, the composition of these two liquids was identical. We chose to investigate a vehicle (50:50 PG:VG) that is commonly used by e-cigarette users. To avoid potential confounding effects, we also elected to use a vehicle that did not contain flavouring since the additives in e-cigarette flavourings induce significant oxidative and inflammatory responses in lung cells and tissues.7 This study observed no deleterious effects of the vehicle, but the authors cannot rule out the potential long-term side effects of components in a wide variety of commercially available e-liquids. Indeed, recent e-cigarette studies found substantial detectable levels of toxic metals, such as aluminium, copper, zinc, lead and chromium.40
In summary, these results show the adverse lung health effects of chronic e-cigarette exposure. In addition, they establish that nicotine is the key component that causes airway hyper-reactivity and lung tissue destruction. e-Cigarettes contain far fewer carcinogens and are less likely to promote carcinogenesis. Thus, though e-cigarettes may have a better safety profile, refraining from nicotine inhalation will likely impact on preserving lung health.
Acknowledgments
The authors thank Dr Moro Salifu and Dr Gerard Turino and the James P. Mara Center for Lung Disease and Ruth Unger and the Mah Jongg Foundation League for their support and guidance. They also acknowledge Michael Myerburg (University of Pittsburgh) for making meniscus scanning software for airway surface liquid volume estimation available.
References
Footnotes
IG-A and PG contributed equally. MS and RF shared senior authorship.
Contributors IG-A and PG: responsible for study design and execution, data collection and manuscript preparation. NB, MC, AJD, BJ, NC, AG and EE: responsible for study execution. MS: responsible for study design and execution and manuscript preparation. RF: responsible for study design, execution, data collection and overall manuscript preparation. He is the guarantor.
Funding This work was supported by grants made available to RF (Flight Attendant Medical Research Institute YCSA 24039, CIA 074047 and the US National Institutes of Health 1R01HL098528-05), MS (Flight Attendant Medical Research Institute, CIA 130033 and the James and Esther King Florida Biomedical Research Program grant#5JK02), PG (Flight Attendant Medical Research Institute YCSA 113380) and IG-A (Flight Attendant Medical Research Institute YCSA 13005).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.