Article Text

Abstract
Background: To investigate the frequency of cardiac troponin I (cTnI) increases in patients with pulmonary embolism (PE) and to assess the correlation between this finding, the clinical presentation, and outcomes.
Methods: Consecutive patients admitted to the coronary care unit with acute PE were prospectively enrolled between January 2000 and December 2001. cTnI was sequentially determined. Various cut off concentrations were analysed, but patients were categorised prospectively as having increased or no increased cTnI based on a cut off concentration of 0.6 ng/ml. The main outcome measure was in-hospital mortality.
Results: On admission, 14 of the 48 patients (29%) had cTnI concentrations greater than the receiver operating characteristic curve value used to diagnose acute myocardial infarction (> 0.6 ng/ml). Subsequently, six patients developed increases for an overall prevalence of 42% (20 of 42). The prevalence was higher when lower cut off concentrations were used: 73% (35 of 48) at the 99th centile and 60% (29 of 48) at the 10% coefficient of variability. Increased cTnI > 0.6 ng/ml was associated with a slower oxygen saturation (86 (7)% v 93 (4)%, p < 0.0001) and more frequent involvement of the main pulmonary arteries as assessed by spiral computed tomography (100% v 60%, p = 0.022). In-hospital mortality was 36% (5 of 14) of patients with increases > 0.6 ng/ml v 3% (1 of 42) of patients with lower concentrations (p = 0.008). Increased cTnI > 0.6 ng/ml on admission was the most powerful predictor of mortality (p = 0.046).
Conclusions: In high risk patients with acute PE, cTnI was frequently detected on admission. It was the strongest independent predictor of mortality.
- troponin I
- pulmonary embolism
- prognosis
- CT, computed tomography
- cTnI, cardiac troponin I
- cTnT, cardiac troponin T
- PE, pulmonary embolism
Statistics from Altmetric.com
Cardiac troponin I (cTnI) is a highly sensitive and specific marker for myocardial cell injury.1 Although raised cTnI concentration in the setting of acute ischaemia has a key role in risk stratification of patients with acute coronary syndromes,2 its occurrence has increasingly been reported in other clinical conditions as well.1,3–5 Among them, acute pulmonary embolism (PE) is a potentially fatal condition for which prompt recognition, risk assessment, and appropriate treatment must be implemented to reduce mortality and morbidity.6 Since the original report on the use of cardiac troponin T (cTnT) concentration as a marker,4 several groups have reported that increases in cTnI concentration are also seen in patients with acute PE.7–9 Only one study, which used a high cut off concentration7 (the concentration determined for acute myocardial infarction from receiver operating characteristic curve), defined its prognostic potential. The others, which used lower cut off concentrations8,9 such as the 10% coefficient of variability and the 99th centile, were interested in right ventricular dysfunction or an explanation for cTnI increases themselves. Furthermore, patients with known coronary artery disease were not excluded from these studies, making interpretation more difficult. Accordingly, this prospective clinical study was designed to assess the prevalence of cTnI positivity at various cut off concentrations and to evaluate the impact of increases on prognosis in a cohort of patients without known coronary artery disease.
PATIENTS AND METHODS
Consecutive patients with acute PE at the Vicenza Hospital, Italy, were enrolled in this study between January 2000 and December 2001. Patients were identified by urgent referral to our coronary care unit with the presumptive diagnosis of acute PE. The study was approved by the institutional review committee of the hospital and under their aegis verbal consent was obtained from all participants to allow for additional blood samples to be obtained. Patients with a history of coronary artery disease (that is, documented angina, previous myocardial infarction, coronary angioplasty, or bypass surgery) were excluded.
A blood sample for determination of cTnI was obtained from patients upon arrival at the coronary care unit together with baseline routine blood tests. Biochemical analysis of cTnI was performed by experienced personnel unaware of the patient’s clinical status. Additional samples were obtained at 8, 16, and 24 hours. The method used was the one step enzyme immunoassay done on the Dimension RXL system (Dade Behring, Newark, Delaware, USA). The upper limit of the reference range for normal healthy adults—that is, the 99th centile of the normal range—is < 0.07 ng/ml. The concentration where the coefficient of variability is 10% is 0.14 ng/ml.10 The suggested concentration for detection of myocardial damage is 0.6 ng/ml.6 A 12 lead ECG was obtained on admission and repeated as needed. Patients were excluded if the ECG was considered to indicate coronary artery disease. An echocardiogram was obtained from as many patients as feasible. Right ventricular involvement was diagnosed if the echocardiogram had evidence of right ventricular enlargement (M mode diameter exceeding 30 mm), paradoxical septal motion, an embolus observed in transit, or tricuspid regurgitation with peak velocity > 2.5 m/s, as suggested by Lualdi and Goldhaber.11 Patients with regional dysfunction suggestive of ischaemic heart disease were excluded. PE was diagnosed by spiral computed tomography (CT), ventilation perfusion lung scanning, pulmonary angiography, or angiomagnetic resonance. Arterial blood gas was analysed before oxygen supplementation. Oxygen saturation and oxygen partial pressure were measured.
Patients were administered O2 and heparin and inotropic agents as clinically indicated. Assisted ventilation was required for patients in shock and with severe hypoxaemia. A thrombolytic agent was administered according to the physician’s assessment of the severity of PE and was independent of the results of cTnI testing. It is the policy in our centre to treat patients with thrombolysis if they have shock or evidence of right ventricular dysfunction on echocardiography.
Data are presented as mean (SD) for continuous variables and as frequencies for categorical variables. Groups were compared by a two tailed unpaired Student’s t test or χ2 statistic with Fisher’s correction when appropriate. Independent predictors of mortality were defined with multivariate logistic regression analysis. By prospective decision, cTnI increase was considered both as a dichotomous (positive versus negative, at a concentration of 6 ng/ml) and a continuous variable. The results of multivariate logistic regression analysis are reported as odds ratios and 95% confidence intervals. A probability value of p < 0.05 was considered significant. All calculations were done with SPSS system 10.0 (SPSS Inc, Chicago, Illinois, USA).
RESULTS
Table 1 shows the baseline characteristics of the study population of 48 patients and table 2 shows their haemodynamic, ECG, and echocardiographic findings. The diagnosis of PE was confirmed mostly by spiral CT (n = 42) or on high probability ventilation perfusion images (n = 4). For two critically ill patients the diagnosis was based on clinical signs and echocardiographic data alone.
Clinical characteristics and relevant findings at presentation in the overall study cohort (n = 48)
Clinical findings on admission according to the presence (cTnI+) or absence (cTnI−) of an early increase in cardiac troponin I (cTnI)
On admission, cTnI was increased above 0.6 ng/ml in 14 of 48 (29%) patients and was below the selected cut point in the remaining 34 (71%). Figure 1 illustrates the change in cTnI concentrations over time in those with increases on admission. An additional six patients had increased cTnI concentrations in the second blood samples drawn eight hours after admission to the coronary care unit. Thus, the overall prevalence of increased cTnI concentrations at this cut off concentration was 20 of 48 (42%).

Time course of cardiac troponin I (cTnI) concentrations in patients with a positive assay on admission. Three of the patients who died during hospitalisation (+) had only one cTnI determination and two had two assays. The remainder were monitored up to 24 hours.
There was no correlation between the duration of symptoms and a positive cTnI. The mean cTnI concentration on admission was 1.5 (0.8) ng/ml in cTnI positive patients and the mean concentration at peak was 1.9 (1.2) ng/ml. The group with increased cTnI on admission had faster heart rates, more frequent echocardiographic right ventricular dilatation/hypokinesia, higher pulmonary artery pressures, and lower oxygen saturations. There was more severe involvement of the main pulmonary arteries (table 3) in patients with increased cTnI concentrations (p = 0.022). Five patients had shock and four of them required endotracheal intubation. cTnI concentrations were increased (> 0.6 ng/ml) in three patients on admission and in one in the second sample.
Spiral computed tomography findings
Thirty two of the 48 (67%) patients met our criteria for the use of thrombolytic agents and were treated with alteplase (tPA) 100 mg over two hours. Of these 32 patients, eight (25%) had increased cTnI and 24 (75%) did not (p = 0.369). All six of the patients who developed increased cTnI were in this group. Overall, there were six in-hospital deaths, a 12.5% in-hospital mortality rate. Five of the patients who died had an increased cTnI concentration on admission and one did not on admission but had an increase at eight hours (five of 14 (36%) v one of 42(3%), p = 0.008). This latter patient had systemic non-Hodgkin’s lymphoma and died of coagulopathy 22 days after PE, without evidence of embolic recurrence. The other five patients died of refractory shock while sill intubated. The mean concentration of cTnI on admission of patients who subsequently died was 2.1 (1.4) ng/ml, significantly higher (p = 0.011) than that of patients with an increased cTnI who survived (0.9 (0.6) ng/ml).
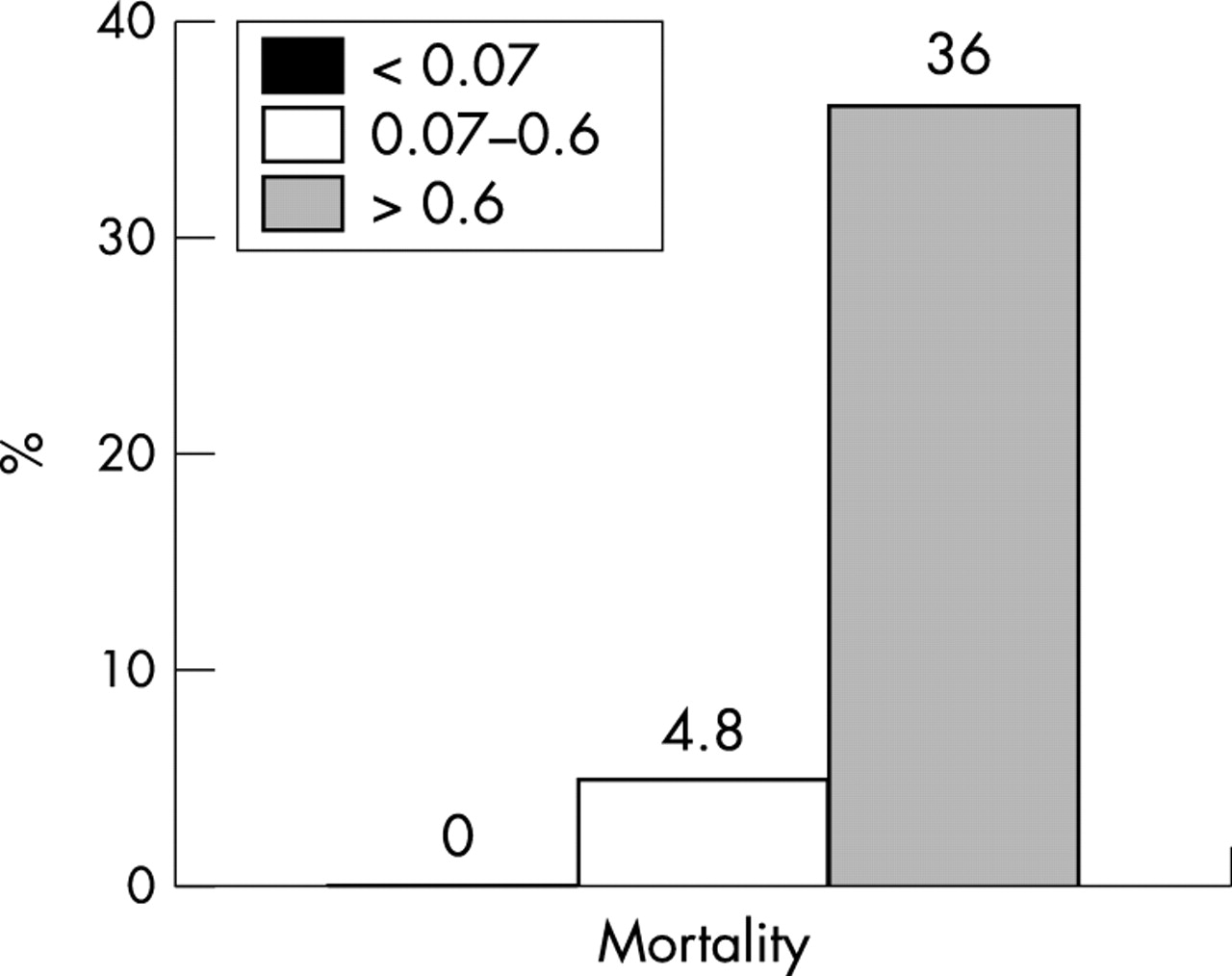
Because even trace amounts of cTnI seem to have prognostic implications for patients with acute coronary syndromes,8 we performed a post hoc analysis of our data looking at patients with cTnI concentrations exceeding the cut point of 0.07 ng/ml. With this cut off concentration, an additional 21 patients (44%) were classified as having increases on admission. On the basis of this criterion, 35 of 48 (73%) patients had increases on admission. When the cut off value at the 10% coefficient of variability was used, as recently suggested,10 29 of 48 patients (60%) had increased concentrations. Mortality increased in a stepwise fashion (fig 2), paralleling the increase of cTnI concentrations (0% for patients with normal cTnI concentrations, 4.8% for patients with modest increases (0.07–0.6 ng/ml), and 36% for those exceeding 0.6 ng/ml; p for trend = 0.005).
{kind=link}
{kind=link}
Relation between cTnI concentrations (< 0.07 ng/ml, 0.07–0.6 ng/ml, and > 0.6 ng/ml) on admission and mortality (%).
Table 4 shows predictors of in-hospital mortality. None of the baseline clinical characteristics—age, sex, previous PE, need for respiratory support or inotropic agents, or use of thrombolysis—was a significant predictor of outcome. Similarly, electrocardiographic or echocardiographic findings were not associated with mortality. While a faster heart rate, a lower oxygen saturation, and a higher pulmonary pressure were all predictors of mortality with univariate analysis, only a positive cTnI on admission > 0.6 ng/ml and any troponin increase (data not shown) maintained a significant independent predictive value in a multivariate model when analysed either as a dichotomous (p = 0.046, odds ratio (OR) 17.9, 95% confidence interval (CI) 1.06 to 303.8) or as a continuous variable (p = 0.007, OR 9.27, 95% CI 1.82 to 47.1).
Predictors of in-hospital mortality
DISCUSSION
Our data confirm and extend the results of prior studies of patients with acute PE. Increased troponin concentration with a high cut off was the most highly predictive factor associated with mortality in this cohort of critically ill patients.7 The six patients who died had an increased concentration and five of them had increases at the time of presentation. Smaller increases and the other later increases were associated with more severe haemodynamic impairment but not with death.7–9 In addition, the frequency of troponin increases in our patients who are unlikely to have ischaemic heart disease was 42% with the higher cut off concentrations and correlated with prognosis: 73% for cut off concentrations advocated by some for the diagnosis of acute myocardial infarction for patients with acute ischaemic heart disease12 and 60% for patients with admission concentrations at the 10% coefficient of variability level, also suggested by some.10 This high frequency of increased cTnI suggests that PE should be considered when patients present with chest pain or shortness of breath and increased concentrations of cTnI. Finally, the early serial concentrations we obtained suggest but do not prove that the rapid release and clearance of troponin described for cTnT also are likely to apply to cTnI.
Prediction of mortality in acute PE is a complex task in which the clinician has to consider the severity of pulmonary vascular obstruction,6 the presence of right ventricular involvement,13 signs of cardiogenic shock,14 the risk of recurrence,15 and baseline adverse characteristics of the patient,16 such as advanced age, cancer, and previous PE. In our series, although only six patients died, none of these commonly used clinical parameters predicted in-hospital mortality. Only cTnI at high cut off concentrations could make that distinction. This finding also held when considering cTnI as a continuous variable, suggesting a quantitative grading of increasing risk. No patients with concentrations below 0.06 ng/ml died, and mortality was 4.8% among patients with slightly increased concentrations (> 0.07 but < 0.6 ng/ml) on admission and 36% among patients with still higher concentrations. These data suggest that initial troponin concentrations can be used to define risk on admission. Patients with high concentrations are clearly at great risk and patients with modest increases deserve additional testing to establish risk more completely. These data are consistent with those of Konstantinides and colleagues,7 who reported that both cTnT and cTnI, with high cut off concentrations for both, predicted in-hospital events in a cohort of 106 patients with acute PE. Patients in our series were older (64 v 61 years) and had a higher prevalence of right bundle branch block on the ECG (42% v 18%) and more frequent echocardiographic right ventricular abnormalities (83% v 44%). Not surprisingly, this profile was associated with a greater overall in-hospital mortality (12.5% v 6.6%) despite much more aggressive use of thrombolysis (67% v 15%). This occurred in part because our study focused only on patients who required acute coronary care unit admission. However, the profile was obtained despite our attempt to exclude patients with underlying coronary artery disease, confirming the potent prognostic effects of cTnI increases in patients with acute PE alone.
The mechanism of cTnI release in PE remains speculative. Increased right ventricular wall stress and low cardiac output with low coronary perfusion pressure in combination are recognised pathophysiological changes that occur with significant PE.11 These mechanisms create a vicious cycle eventually resulting in ischaemia to the right ventricle. In this model, the acutely stressed right ventricular myocytes are the origin of cTnI release. This concept is supported by the pathological findings by Jerjes-Sanchez and colleagues,17 who found evidence of right ventricular infarction in the absence of coronary artery disease in four patients who died of massive PE. This mechanism is more likely in our patients, in that we attempted to exclude prior coronary artery disease. We cannot exclude the presence of a previously silent coronary abnormality.
Our data show that increased cTnI concentration is associated with more severe disease. Increases were associated with significantly more extensive and proximal involvement of the pulmonary vasculature, as assessed by spiral CT, and many of the clinical features of these patients (oxygen saturation, blood pressure, heart rate, echocardiographic right ventricular abnormalities, and estimated pulmonary artery pressure) also suggested a more severe clinical profile. Thus, all these findings support the concept that there is a link between the degree of troponin increase and the severity of the clinical presentation. Available clinical data,4,7–9 including the present report, suggest that increases in the cardiac troponins portend an adverse prognosis for patients with acute PE, as well as for patients with acute coronary syndromes.2 Risk stratification can be complex and it is unlikely that cTnI concentration will provide perfect prognostication given the complexity of clinical care, but it obviously is a potent risk marker. Further investigation is required to verify whether risk stratification based on cardiac troponin concentrations may translate into targeted treatment strategies. In addition, it is apparent, given the potential commonality of presentation of acute PE and acute coronary syndromes and the possibility of conjoint presentation, that PE should be considered in patients who present with compatible symptoms and increased concentrations of cTnI.
We did not characterise the kinetics of cTnI release and clearance but it did appear from the data obtained during the first 24 hours (fig 1) that they were similar to those described for cTnT, where increases occur rapidly but do not persist beyond 40 hours.18
This report concerns patients with a severe clinical profile that warranted admission to an intensive care unit, rather than an unselected series. That is why thrombolytic treatment was used so liberally. It may well be that more aggressive use of such treatment would improve prognosis, but this study was too small and treatment was at the discretion of the treating physicians and not randomised. Thus, the benefits of thrombolysis may well have been missed. We believe that a trial of thrombolysis guided by troponin concentration is warranted. We also excluded patients with known coronary artery disease. Thus, the principles derived from this study and from others may need to be moderated in the more complex situation of clinical care where no one is excluded.
In conclusion, cTnI is frequently detected in the first hours after admission in patients with confirmed acute PE and is associated with an adverse clinical profile. cTnI increase is a powerful independent predictor of mortality and perhaps would be still better if combined with other markers such as brain natriuretic peptide.19 Acute PE should be one of the diagnoses considered whenever critically ill patients have increased cTnI concentrations and ischaemic heart disease seems less likely.
Acknowledgments
The authors acknowledge, with appreciation, the secretarial assistance of Ms Jennifer Bolstad.
REFERENCES
Footnotes
-
Dr Allan Jaffe is a consultant for Dade Behring as well as for Roche and Beckman Coulter, all companies that make troponin assays. There are no other conflicts of interest.