Article Text

Abstract
Background Non-alcoholic fatty liver disease is an obesity-related chronic liver disorder ranging from simple steatosis to non-alcoholic steatohepatitis (NASH), which may progress to liver fibrosis and cirrhosis.
Objective Tto investigate the role of Toll-like receptor (TLR) 4 in mediating the transition from steatosis to inflammation.
Methods ApoE−/−/TLR4mut mice and ApoE−/−/TLR4 wild-type mice (ApoE−/−/TLR4-WT) were generated by cross-breeding an ApoE-deficient (ApoE−/−) strain with TLR4-mutant (TLR4mut) mice, which were fed with high-fat, high-cholesterol (HFHC) diet to induce obesity.
Results ApoE−/−/TLR4-WT mice fed with an HFHC diet for 12 weeks developed typical pathological features of NASH, which is associated with obesity and the metabolic syndrome. By contrast, ApoE−/−/TLR4mut mice lacking functional TLR4 were resistant to HFHC diet-induced liver inflammation and injury and were less susceptible to the diet-induced production of reactive oxygen species (ROS) and proinflammatory cytokines. In ApoE−/−/TLR4-WT mice, X-box binding protein-1 (XBP-1), a transcription factor involved in the unfolded protein responses, was activated in the liver by an HFHC diet, whereas XBP-1 activation was abrogated in ApoE−/−/TLR4mut mice. In primary rat Kupffer cells, endotoxin induced XBP-1 activation through ROS production, whereas siRNA-mediated knockdown of XBP-1 expression resulted in a marked attenuation in endotoxin-evoked NF-κB activation and cytokine production. Furthermore, adenovirus-mediated expression of dominant negative XBP-1 led to a significant attenuation in HFHC diet-induced liver inflammation and injury in mice.
Conclusions These findings support the key role of TLR4 in Kupffer cells in mediating the progression of simple steatosis to NASH, by inducing ROS-dependent activation of XBP-1.
- Obesity
- inflammation
- fatty liver disease
- oxidative stress
- metabolic bone disease
- liver
- DNA damage
- drug development
- drug induced liver injury
- drug resistance
- drug toxicity
- fatty liver
- nonalcoholic steatohepatitis
- cell biology
Statistics from Altmetric.com
- Obesity
- inflammation
- fatty liver disease
- oxidative stress
- metabolic bone disease
- liver
- DNA damage
- drug development
- drug induced liver injury
- drug resistance
- drug toxicity
- fatty liver
- nonalcoholic steatohepatitis
- cell biology
Significance of this study
What is already known about this subject?
Obesity-related non-alcoholic fatty liver disease/non-alcoholic steatohepatitis (NAFLD/NASH) is closely associated with increased circulating endotoxin and fatty acids.
Toll-like receptor (TLR) 4 serves as a receptor for both endotoxin and fatty acids and has been linked to endoplasmic reticulum (ER) stress.
X-box binding protein-1 (XBP-1) is a key transcription factor that mediates the unfolded protein responses in ER stress.
TLR4 null mice are less susceptible to chemical-induced liver fibrosis and fructose-induced hepatic steatosis.
What are the new findings?
ApoE-deficient/TLR4 wild-type (ApoE−/−/TLR4-WT) mice fed with a high-fat, high-cholesterol (HFHC) diet develop typical pathological features of NASH, which is associated with obesity and the metabolic syndrome.
TLR4 expression is selectively elevated in hepatic macrophages in ApoE−/−/TLR4-WT mice fed an HFHC diet.
TLR4 mediates the transition of benign steatosis to steatohepatitis by reactive oxygen-dependent activation of XBP-1, thereby leading to NF-κB activation and proinflammatory cytokine production.
Inactivation of either TLR4 or XBP-1 protects against HFHC diet-induced steatohepatitis and liver injury in ApoE−/− mice.
How might it impact on clinical practice in the foreseeable future?
In light of the obligatory role of XBP-1 in TLR4-induced liver inflammation and injury, therapeutic interventions targeting the TLR4/XBP-1 signalling cascade may be a promising strategy for treating NAFLD and NASH.
Introduction
Non-alcoholic fatty liver disease (NAFLD) comprises a spectrum of disorders ranging from simple steatosis to non-alcoholic steatohepatitis (NASH), fibrosis and cirrhosis.1 NAFLD and NASH coexist with a cluster of obesity-related cardio-metabolic complications, including insulin resistance, type 2 diabetes, dyslipidaemia and atherosclerosis and are considered to be the hepatic manifestation of the metabolic syndrome.2 3 Owing to the rapid rise in the prevalence of obesity and metabolic syndrome, NAFLD has become the most common cause of chronic liver diseases worldwide.
Simple steatosis has a benign clinical course, whereas patients with NASH can develop fibrosis, cirrhosis and complications such as hepatocellular carcinoma.4 Therefore, the progression of hepatic steatosis to steatohepatitis is the crucial step in the development of obesity-related NASH. The ‘two-hit’ model originally proposed by Day and James suggests that after a first hit by hepatic steatosis and insulin resistance, another hit is needed to develop NASH and ultimately, fibrosis.5 In obesity, many parallel hits derived from both gut and adipose tissue, including the endotoxin (lipopolysaccharide (LPS)), proinflammatory adipokines and toxic lipids, may contribute to liver inflammation.3 6 However, the cellular and molecular events underlying the transition of bland steatosis towards a more progressive, inflammatory disease phenotype in the liver remain poorly characterised.
Toll-like receptors (TLRs) are members of the pattern-recognition receptor superfamily that are of central importance during the host defence against invading pathogens.7 A growing body of evidence suggests that TLRs, especially TLR4, have a key role in the pathogenesis of chronic inflammatory liver diseases.6 TLR4 is the principal receptor for endotoxin, which is a central mediator of liver inflammation associated with both alcoholic and non-alcoholic liver disease.6 The close association between NAFLD/NASH and increased circulating endotoxin has been seen in both obese animals and humans.8 9 The interaction of TLR4 with LPS results in liver injury and fibrosis by triggering the production of a myriad of proinflammatory cytokines6 and by inducing oxidative stress.10 Besides endotoxin, TLR4 is also activated by free fatty acids, which is also an important factor contributing to obesity-induced liver necroinflammation.
Several previous studies in TLR4 mutant (TLR4mut) mice have demonstrated an aetiological role for TLR4 in the pathogenesis of chronic inflammatory liver diseases.11–14 C3H/HeJ TLR4mut mice, which lack functional TLR4, strongly reduce alcohol-induced liver injury11 and are also resistant to carbon tetrachloride-mediated hepatic fibrosis.13 Furthermore, inactivation of TLR4 results in a marked attenuation of steatohepatitis induced by a methionine-/choline-deficient diet,12 and also leads to a significant decrease in fructose-induced hepatic steatosis.14 In obese mice, TLR4 mediates inflammation in adipose tissue, which in turn induces systemic insulin resistance.15 However, owing to a lack of suitable animal models which fully recapitulate the natural history of obesity-induced NAFLD/NASH, the precise pathophysiological function of TLR4 signalling in the development of this disease remains poorly understood.
ApoE-deficient mice (ApoE−/−) fed with Western-style atherogenic diet have been widely used as a rodent model for atherosclerosis.16 A number of studies have shown that ApoE-null mice may develop the full-spectrum of liver pathology that characterises human NASH in an appropriate metabolic context similar to obesity.17 18 In this study, we further validated the feasibility of using ApoE−/− mice receiving a high-fat, high-cholesterol (HFHC) diet as a rodent model for obesity-induced NASH and investigated the role of TLR4 signalling and its cross-talk with oxidative stress and endoplasmic reticulum (ER) stress in the pathogenesis of diet-induced NASH by crossing ApoE−/− mice with the TLR4mut mice.
Materials and methods
Animal studies
C57 BL/6J ApoE−/− mice and C3H/HeJ TLR4-mutant (TLR4mut) mice, which bear a non-functional mutation at histidine-712, were obtained from the Jackson Laboratory. C3H/HeJ TLR4mut mice were back-crossed into C57 BL/6J ApoE−/−mice for more than 10 generations to obtain C57 BL/6J ApoE−/− mice bearing either TLR4 wild-type or TLR4 mutant genotype. These mice were fed with either standard chow or an HFHC diet (D12079B, Research Diet, New Brunswick, New Jersey, USA) containing 41% fat, 17% protein, 43% carbohydrate and 0.21% cholesterol for 12 weeks. All animals were kept under 12 h light–dark cycles at 22–24°C, with free access to water. A glucose tolerance test was conducted as we previously described.19 All animal experimental procedures were approved by the Committee on the Use of Live Animals for Teaching and Research of the University of Hong Kong and were carried out in accordance with the Guide for the Care and Use of Laboratory Animals.
Detection of XBP-1 splicing by RT-PCR
The X-box binding protein-1 (XBP-1) mRNA splicing in mouse livers and rat hepatic macrophages (Kupffer cells) was analysed using RT-PCR as described previously.20 The sequences of primers used for PCR amplification of mouse and rat XBP-1 are listed in online supplementary table 1. The PCR products were resolved by electrophoresis on a 5% polyacrylamide gel and visualised by ethidium bromide staining.
Isolation and culture of Kupffer cells and hepatocytes
Primary Kupffer cells and hepatocytes from Wistar rats and ApoE−/−/TLR4-WT mice were prepared by liver perfusion, as detailed in supplementary materials and methods online.
Analysis of NF-κB activation
Primary rat Kupffer cells were cultured to confluence in 60 mm culture dishes and were then incubated with LPS for different periods. Nuclear and cytoplasmic extracts were prepared using mammalian nuclear protein extraction reagent (Pierce, Rockford, Illinois, USA). Activation of the NF-κB p50 subunit in the nuclear extracts was detected in nuclear protein extracts using a commercial kit (EZ-Detect Transcription Factor Kit; Pierce).
Statistical analysis
Experiments were performed routinely using five to seven mice per group with values presented as means±SEM. Statistical significance was determined by one-way analysis of variance or Student t test using the Statistical Package for Social Sciences V.14.0 (SPSS). In all statistical comparisons, a p value <0.05 was used to indicate a significant difference.
Additional methods
Detailed methodology is described in the online supplementary ‘Materials and methods’ section.
Results
TLR4 inactivation protects against high-fat, high-cholesterol (HFHC) diet-induced liver injury in ApoE−/− mice
C57 BL/6J ApoE−/− mice fed with an HFHC diet are commonly used rodent models for atherosclerosis.16 A growing body of evidence suggests that atherosclerosis and NAFLD are closely related inflammatory disorders.21 Inactivation of TLR4, a central regulator of innate immunity and inflammation, prevents atherosclerotic plaque formation in ApoE−/− mice.22 Therefore, we investigated the potential roles of TLR4 in the pathogenesis of NAFLD in ApoE−/− mice. To this end, C3H/HeJ TLR4mut mice were back-crossed into C57 BL/6J ApoE−/− mice for at least 10 generations to generate ApoE−/−/TLR4mut mice and ApoE−/−/TLR4 wild-type (TLR4-WT) mice. Histological analysis showed that ApoE−/−/TLR4-WT mice fed with an HFHC diet for 12 weeks exhibited typical pathological features of NASH, including macrovesicular and microvesicular steatosis, hepatocellular ballooning and a component of polymorphonuclear leucocytes indicative of lobular inflammation (figure 1A). Semiquantification analysis of steatohepatic lesions using the NASH Clinical Research Network (NASH CRN) scoring system23 demonstrated an approximately fivefold increase in NASH scores in ApoE−/−/TLR4-WT mice fed with an HFHC diet compared with those fed with standard chow (SC) (figure 1B). In addition, ApoE−/−/TLR4-WT receiving an HFHC diet displayed an approximately twofold increase in serum levels of alanine aminotransferase (ALT; figure 1C). Noticeably, these pathological changes were also accompanied by the development of obesity and its related metabolic syndromes, including increased body weight, impaired glucose tolerance and atherosclerosis (supplementary figure 1). Taken together, these findings suggest that ApoE−/−/TLR4-WT mice fed with an HFHC diet develop symptoms typical of obesity-induced NASH similar to those in human beings.
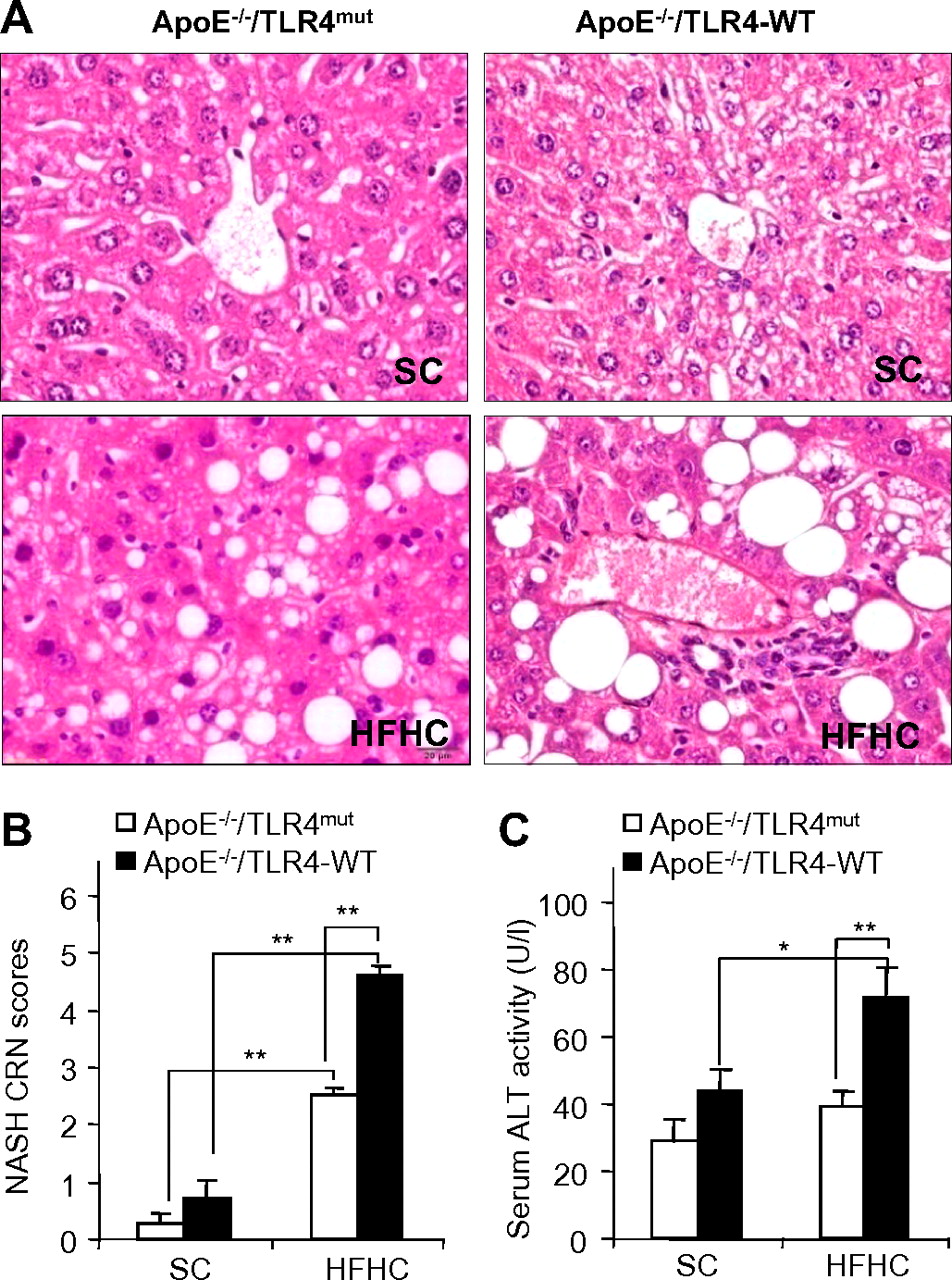
Effect of Toll-like receptor 4 (TLR4) inactivation on high-fat, high-cholesterol (HFHC) diet-induced liver injury in mice. ApoE−/−/TLR4-WT and ApoE−/−/TLR4mut mice were fed with standard chow (SC) or HFHC diet for 12 weeks. WT, wild-type. (A) Representative images of haematoxylin and eosin-stained sections of liver tissues (original magnification: ×400). (B) Histological non-alcoholic steatohepatitis (NASH) scores determined according to the NASH Clinical Research Network (CRN) scoring system. (C) Liver injury as measured by serum alanine amino transferase (ALT) activity. Data are expressed as mean±SEM, n=5–6. *p<0.05, **p<0.01. This figure is produced in colour in the online journal—please visit the website to view the colour figure.
Compared with the changes seen in ApoE−/−/TLR4-WT mice, HFHC diet-induced steatosis, inflammatory infiltration and hepatocellular ballooning were markedly attenuated in ApoE−/−/TLR4mut mice (figure 1A). Accordingly, NASH CRN scores in ApoE−/−/TLR4mut mice were more than 50% lower than those in ApoE−/−/TLR4-WT mice (figure 1B). These histological improvements in ApoE−/−/TLR4mut mice were accompanied by an over 60% reduction in serum ALT levels (figure 1C).
Analysis of hepatic lipid contents by both Oil Red O staining of liver sections and biochemical analysis showed a modest, but significant reduction in HFHC diet-induced lipid accumulation in ApoE−/−/TLR4mut mice compared with ApoE−/−/TLR4-WT mice (supplementary figure 2A,B). Sirius red staining showed that ApoE−/−/TLR4-WT mice and ApoE−/−/TLR4mut mice receiving SC or an HFHC diet did not develop liver fibrosis (supplementary figure 3A). In ApoE−/−/TLR4-WT mice, macrophage infiltration in adipose tissue, as measured by FACS analysis using anti-CD11b and anti-F4/80 antibodies, was significantly increased by feeding an HFHC diet, whereas such an HFHC diet-induced change was significantly attenuated in ApoE−/−/TLR4mut mice (supplementary figure 3B).
TLR4 expression is selectively raised in hepatic macrophages in HFHC diet-induced NASH
Several previous studies have reported an increased expression of TLR4 gene in muscle24 and monocytes25 in obese and type 2 diabetic subjects. Therefore, we next investigated whether hepatic TLR4 expression was altered in HFHC diet-induced NASH. Quantitative real-time PCR analysis demonstrated a markedly upregulated TLR4 mRNA expression in liver tissues from both ApoE−/−/TLR4mut and ApoE−/−/TLR4-WT mice (figure 2A). Cell fractionation analysis showed that an HFHC diet-induced increase in TLR4 expression occurred predominantly in non-parenchymal cells (NPCs), whereas no change was seen in parenchymal cells (PCs) (figure 2B). To determine the specific type of cell(s) responsible for HFHC diet-induced increase in TLR4 expression, we further isolated hepatic macrophages (Kupffer cells) from the NPC fraction and compared the levels of TLR4 mRNA expression between Kupffer cells and hepatocytes. This analysis showed that TLR4 expression in Kupffer cells was approximately 20-fold higher than in hepatocytes. An HFHC diet-induced elevation of TLR4 expression was seen in Kupffer cells, but not in hepatocytes (figure 2C). Furthermore, HFHC diet-induced TLR4 expression in the liver was completely abrogated when Kupffer cells were depleted by intraperitoneal injection of gadolinium chloride (GdCl3) (figure 2D), confirming that Kupffer cells are the predominant contributor to the elevated TLR4 expression in HFHC diet-induced NASH in mice.

Toll-like receptor 4 (TLR4) expression is selectively induced in hepatic macrophages by high-fat, high-cholesterol (HFHC) diet. (A) Real-time PCR quantification of TLR4 mRNA levels in liver tissues from ApoE−/−/TLR4-WT and ApoE−/−/TLR4mut mice receiving either standard chow (SC) or an HFHC diet for 12 weeks. WT, wild-type. (B) Protein levels of TLR4 in the parenchymal cell (PC) fraction and non-parenchymal cell (NPC) fraction of ApoE−/−/TLR4-WT mice determined by immunoblotting analysis. (C) The TLR4 mRNA levels in hepatocytes and hepatic macrophages (Kupffer cells) isolated from ApoE−/−/TLR4-WT mice receiving standard chow or an HFHC diet. (D) TLR4 mRNA levels in the liver from ApoE−/−/TLR4-WT mice fed with standard chow, or an HFHC diet with or without Kupffer cells. (Kupffer cells were depleted by intraperitoneal injection of gadolinium chloride (GdCl3).) n=5–7. *p<0.05, **p<0.01.
Inactivation of TLR4 abolishes HFHC diet-induced elevation in hepatic macrophage infiltration and cytokine production
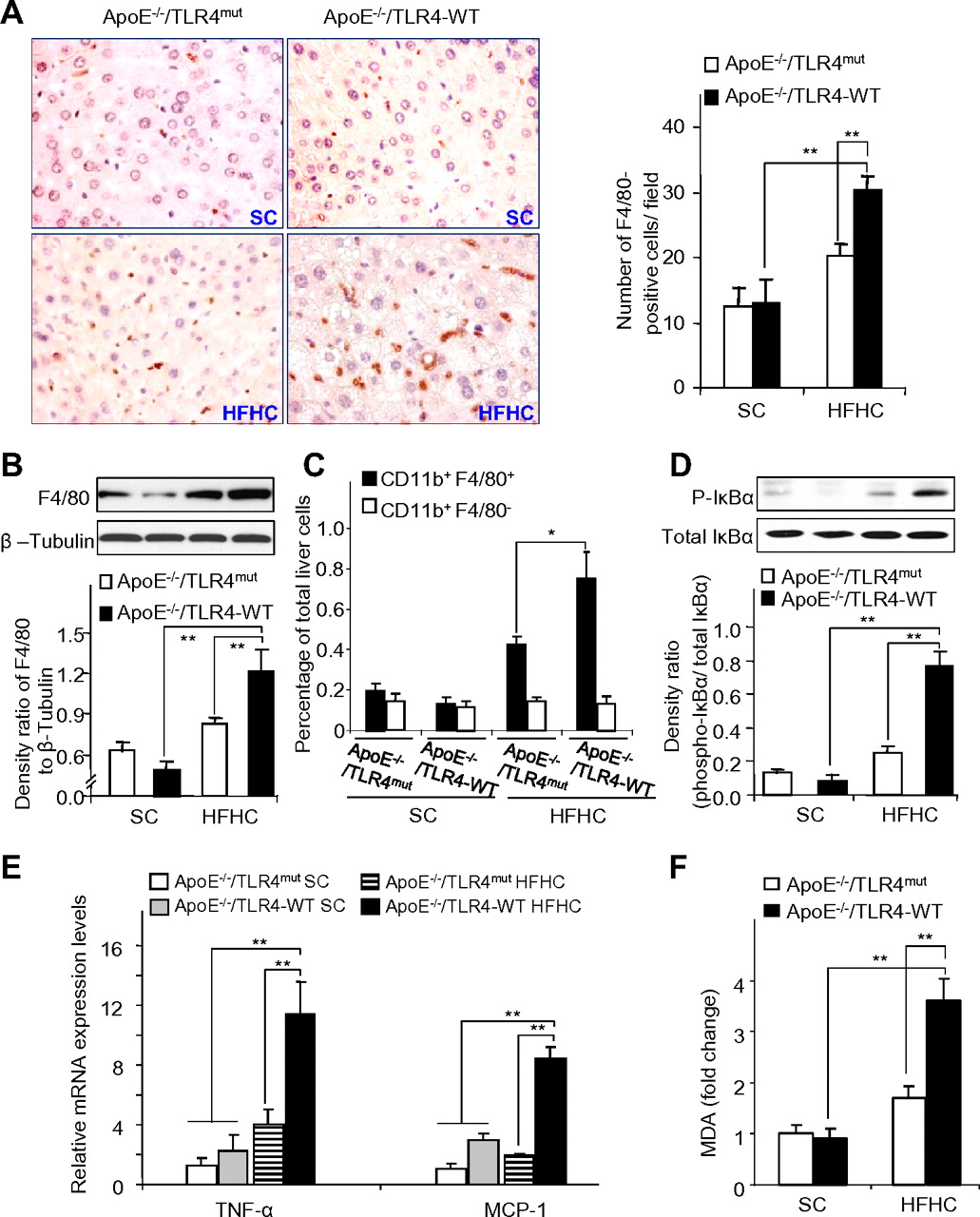
Augmented inflammatory responses are the hallmark of obesity-induced NASH.26 We next investigated whether elevated TLR4 expression contributed to HFHC diet-induced macrophage activation and inflammation. Immunostaining of macrophage marker F4/80 in liver sections showed that HFHC diet feeding led to a significant increase in hepatic macrophage infiltration in ApoE−/−/TLR4-WT mice, whereas this change was compromised in ApoE−/−/TLR4mut mice (figure 3A). Likewise, Western blot data showed that the magnitude of HFHC diet-induced F4/80 protein expression in liver tissues of ApoE−/−/TLR4mut mice was much smaller than that in ApoE−/−/TLR4-WT mice (figure 3B).

Inactivation of Toll-like receptor 4 (TLR4) attenuates high-fat, high-cholesterol (HFHC) diet-induced hepatic macrophage accumulation and inflammation and inhibits the production of reactive oxygen species in mice. (A) Immunohistochemical staining of the macrophage marker F4/80 in liver sections from ApoE−/−/TLR4-WT and ApoE−/−/TLR4mut mice receiving either standard chow (SC) or an HFHC diet for 12 weeks with original magnification at ×400 (left) and quantification of F4/80-positive cells in each field (right). WT, wild-type. (B) Representative immunoblots for F4/80 in liver tissue (upper) and densitometric analysis of F4/80 protein abundance relative to β-tubulin (lower). (C) Quantitative analysis of the percentage of Gr1hi cells (defined as CD11b+F4/80+ cells, black bar) and Gr1lo cells (defined as CD11b+F4/80− cells, open bar) in total liver cells. (D) Representative immunoblots for phosphorylated IκBα in liver tissue (upper) and densitometric analysis of phosphorylated I-κBα protein abundance relative to total IκBα (lower). (E) The mRNA levels of tumour necrosis factor α (TNFα) and monocyte chemoattractant protein-1 (MCP-1) in liver tissues were determined by real-time PCR. (F) Malondialdehyde (MDA) contents in liver tissues from ApoE−/−/TLR4-WT and ApoE−/−/TLR4mut mice receiving SC or an HFHC diet for 12 weeks were expressed as fold changes relative to ApoE−/−/TLR4-WT receiving standard chow; n=5–7. *p<0.05, **p<0.01. This figure is produced in colour in the online journal—please visit the website to view the colour figure.
In line with previous reports,27 28 FACS analysis using the myeloid marker CD11b and the macrophage marker F4/80 antigen identified two distinct subsets of intrahepatic monocytes/macrophages, including CD11b+F4/80+ cells and CD11b+F4/80− cells. CD11b+F4/80+ cells were found to express a high level of Gr1 (Ly6C), whereas CD11b+/F4/80− cells expressed extremely low levels of Gr1, thereby termed Gr1hi and Gr1lo monocytes respectively. Notably, the number of Gr1hi, but not Gr1lo monocytes, was significantly increased in the liver of ApoE−/−/TLR4-WT mice after HFHC diet feeding, whereas such an HFHC diet-induced elevation of Gr1hi was markedly decreased in ApoE−/−/TLR4mut mice (figure 3C). The subpopulations of T cells (CD4, CD8), NK cells (NK1.1) and neutrophils (Ly6G) were modestly elevated in HFHC fed-ApoE−/−/TLR4-WT mice compared with those receiving SC, whereas such HFHC diet-induced changes were less obvious in ApoE−/−/TLR4mut mice (data not shown).
Phosphorylation of IκB-α, a marker of NF-κB activation, was markedly suppressed in liver tissues of HFHC diet-fed ApoE−/−/TLR4mut mice relative to that in ApoE−/−/TLR4-WT mice receiving an HFHC diet (figure 3D). When fed with an HFHC diet, hepatic expressions of tumour necrosis factor α (TNFα) and monocyte chemoattractant protein-1 (MCP-1) were significantly raised in ApoE−/−/TLR4-WT mice compared with those receiving standard chow, whereas such changes were abrogated in ApoE−/−/TLR4mut mice (figure 3E). Taken together, these findings suggest that TLR4 inactivation protects mice from HFHC diet-induced chronic liver injury as well as inflammation.
TLR4 in hepatic macrophages is crucial for HFHC diet-induced NASH in ApoE−/− mice
To further validate the role of TLR4 in hepatic macrophages in mediating NASH development, we performed bone marrow transplantation (BMT) experiments in ApoE−/−/TLR4-WT and ApoE−/−/TLR4mut mice. To this end, bone marrow (BM) cells isolated from either ApoE−/−/TLR4-WT mice or ApoE−/−/TLR4mut mice were transplanted into 6-week-old recipient ApoE−/−/TLR4-WT mice and ApoE−/−/TLR4mut mice that had been lethally irradiated and injected with GdCl3. Genotyping analysis using genomic DNA extracted from blood confirmed over 95% of reconstitution of BM-derived blood cells at 4 weeks after BMT (data not shown). Notably, HFHC diet-induced elevation of NASH CRN scores, serum ALT levels and expression of proinflammatory cytokines were significantly decreased in ApoE−/−/TLR4-WT mice receiving BM cells isolated from ApoE−/−/TLR4mut mice compared with those derived from ApoE−/−/TLR4-WT donors (figure 4A–D). Conversely, ApoE−/−/TLR4mut mice receiving ApoE−/−/TLR4-WT-derived BM cells exhibited markedly enhanced liver lesions and inflammation as compared with ApoE−/−/TLR4mut mice receiving ApoE−/−/TLR4mut mice-derived BM cells, suggesting that hepatic macrophages mediate obesity-associated NASH development in a TLR4-dependent manner.

Bone marrow transplantation (BMT)-mediated Toll-like receptor 4 (TLR4) alteration in hepatic macrophages modifies high-fat, high-cholesterol (HFHC) diet-induced non-alcoholic steatohepatitis (NASH) development in ApoE−/− mice. Six-week-old male ApoE−/−/TLR4-WT and ApoE−/−/TLR4mut mice receiving an HFHC diet were treated with GdCl3 and lethal irradiation (9 Gy) and were then used as recipients to receive transplantation with bone marrow cells obtained from either ApoE−/−/TLR4-WT or ApoE−/−/TLR4mut mice. WT, wild-type. (A) Representative haematoxylin and eosin-stained liver sections with original magnification at ×400. (B) Histological NASH Clinical Research Network (CRN) scores determined as in figure 1. (C) Serum alanine aminotransferase (ALT) activities. (D) Hepatic mRNA expression of tumour necrosis factor α (TNFα; left) and monocyte chemoattractant protein-1 (MCP-1; right) determined by real-time PCR. Data are expressed as mean±SEM, n=5–6. **p<0.01. This figure is produced in colour in the online journal—please visit the website to view the colour figure.
TLR4 mediates HFHC diet-induced accumulation of reactive oxygen species and activation of XBP-1 in the liver
Cellular stresses, including oxidative stress and ER stress, have a key role in promoting the transition from simple steatosis to NASH.29 We next investigated whether oxidative stress and/or ER stress are involved in TLR4-mediated hepatic inflammation induced by HFHC diet. In ApoE−/−/TLR4-WT mice receiving an HFHC diet, intrahepatic levels of malondialdehyde (MDA, a biomarker of lipid peroxidation) were significantly increased compared with those receiving SC (figure 3F). By contrast, the effect of HFHC diet on elevation of hepatic MDA was largely abrogated in ApoE−/−/TLR4mut mice (figure 3F).
In ApoE−/−/TLR4-WT mice receiving an HFHC diet, all the three branches of ER stress-induced unfolded protein response (UPR) pathways were activated, as shown by increased release of the cleaved fragment of activating transcription factor 6 (ATF6,30 (figure 5A), augmented phosphorylation of eukaryotic translational initiation factor 2α (eIF2α) (figure 5B),31 and induced mRNA splicing of XBP-1 (figure 5C). On the other hand, inactivation of TLR4 led to differential changes in HFHC diet-induced UPR pathways. In ApoE−/−/TLR4mut mice, HFHC diet-elicited phosphorylation of eIF2α was further enhanced, whereas activation of ATF6 exhibited no obvious change compared with ApoE−/−/TLR4-WT mice (figure 5A,B). By contrast, HFHC diet-induced activation of XBP-1 was reduced in ApoE−/−/TLR4mut mice (figure 5C). Further cell fractionation analysis demonstrated that suppression of XBP-1 activation and augmentation of eIF2α phosphorylation were seen only in NPCs of HFHC diet fed-ApoE−/−/TLR4mut mice when compared with ApoE−/−/TLR4-WT mice. By contrast, TLR4 inactivation had no obvious impact on HFHC diet-induced activation of the three UPR pathways in PCs (supplementary figure 4A–D).

Differential effects of TLR4 inactivation on the three branches of the unfolded protein response pathways in liver. (A, B) The protein extracts of liver tissues from ApoE−/−/TLR4-WT and ApoE−/−/TLR4mut mice on standard chow (SC) or HFHC diet for 12 weeks were subjected to immunoblotting analysis for ATF6 and its proteolytic fragment ATF6f (A, upper) and for phosphorylated eIF2α (p-eIF2α) (B, upper). The histograms in the lower panel represent the densitometric quantification of ATF6f (A) and p-eIF2α (B) relative to ATF6 and β-tubulin, respectively. (C) RNA isolated from liver tissues was subjected to RT-PCR analysis using primers specific to XBP-1 to detect unspliced XBP-1 (XBP-1u) and spliced XBP-1 (XBP-1s). The ratio of XBP-1s/XBP-1u is shown in the lower panel. n=5–8. *p<0.05, **p<0.01.
LPS-evoked inflammation is dependent on ROS-mediated XBP-1 activation in rat hepatic macrophages
Hepatic macrophages are the primary source of proinflammatory cytokines during the transition from steatosis to NASH.32 Therefore, we next isolated primary macrophages from the liver tissues of Wistar rats to further delineate the roles of reactive oxygen species (ROS) and XBP-1 in TLR4-mediated inflammation. As expected, stimulation of rat hepatic macrophages with the TLR4 agonist LPS induced a markedly increased production of proinflammatory cytokines including TNFα and MCP-1 (figure 6A) and NF-κB activation (figure 6B,C); these changes were accompanied by increased ROS accumulation and XBP-1 mRNA splicing (figure 6D,E). Treatment with the antioxidant N-acetylcysteine or tempol significantly attenuated LPS-induced production of proinflammatory cytokines and activation of NF-κB and also markedly reduced LPS-induced mRNA splicing of XBP-1, suggesting that ROS is the upstream event indispensible for XBP-1 activation by the TLR4 agonist.
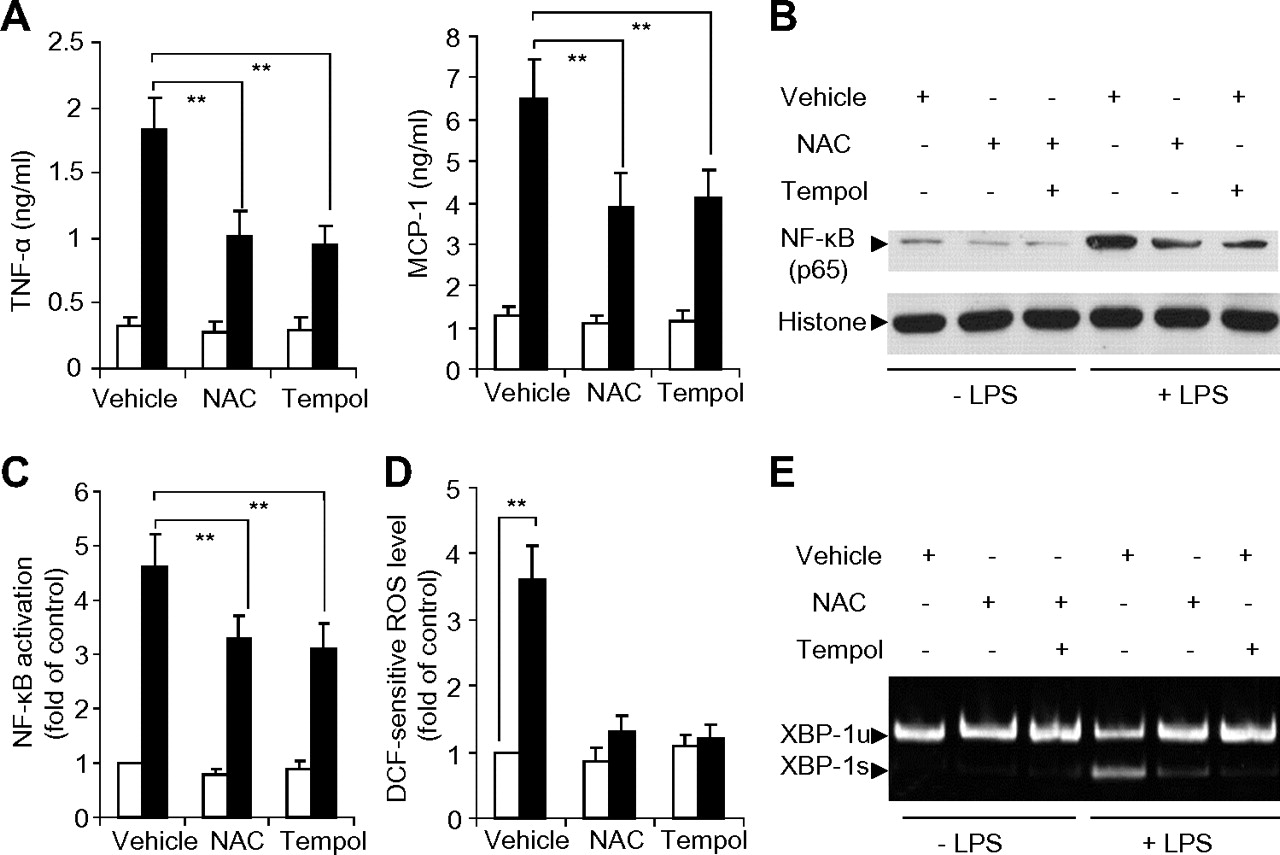
Effects of antioxidants on lipopolysaccharide (LPS)-induced cytokine production, NF-κB activation, reactive oxygen species (ROS) accumulation and XBP-1 mRNA splicing in rat primary Kupffer cells. Kupffer cells isolated from Wistar rats were pre-treated with 2 mM N-acetylcysteine, 1 mM tempol or vehicle for 4 h, followed by stimulation with 50 ng/ml LPS. (A) Protein levels of tumour necrosis factor α (TNFα) and monocyte chemoattractant protein-1 (MCP-1) were quantified by ELISA at 6 h after stimulation with LPS (black bar) or vehicle control (open bar). (B) Representative blot for NF-κB (p65) expression in nuclear extracts at 30 min after LPS stimulation. (C) NF-κB p50 binding activity as measured by ELISA. (D) Intracellular dichlorofluorescein-sensitive ROS concentration at 30 min after LPS stimulation. (E) The mRNA splicing of XBP-1 as determined by RT-PCR at 6 h after LPS stimulation. n=5–8. **p<0.01.
Transfection of rat hepatic macrophages with siRNA specific to XBP-1 for 48 h led to an approximately 72% reduction in XBP-1 expression (figure 7A). LPS-induced production of proinflammatory cytokines and activation of NF-κB were significantly blunted by knocking down XBP-1 expression (figure 7B–E). Taken together, these findings imply that ROS-induced XBP-1 activation is an important signalling event that contributes to TLR4-mediated inflammation in hepatic macrophages.
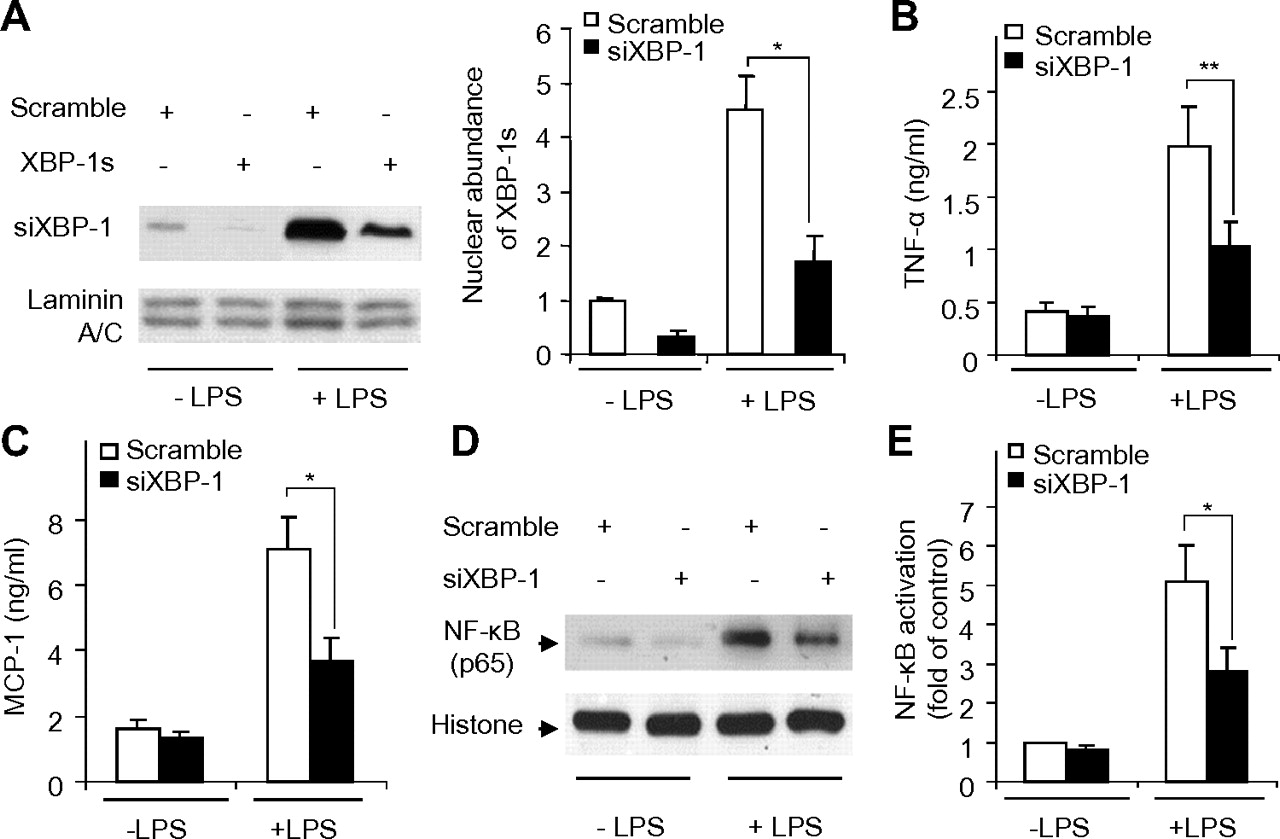
Effects of siRNA-mediated knockdown of XBP-1 on lipopolysaccharide (LPS)-induced cytokine production and NF-κB activation in rat primary Kupffer cells. Cells were transfected with siRNA specific to XBP-1 (siXBP-1) or scrambled control for 24 h, followed by stimulation with 50 ng/ml LPS for another 6 h. (A) Nuclear extracts from rat Kupffer cells transfected with siXBP-1 or scrambled control were subjected to immunoblotting for detection of spliced XBP-1 protein (XBP-1s). The bar chart in the right panel represents the densitometric analysis of XBP-1s abundance relative to the nuclear resident protein laminin A/C. The protein levels of tumour necrosis factor α (TNFα) (B) and monocyte chemoattractant protein-1 (MCP-1) (C) were quantified with ELISA. (D) NF-κB (p65) levels in nuclear extracts detected by immunoblotting analysis. (E) NF-κB p50 binding activity as measured by ELISA. n=5–8. *p<0.05, **p<0.01.
Adenovirus-mediated expression of dominant negative (DN) XBP-1 attenuates HFHC diet-induced liver inflammation in mice
To further delineate the role of XBP-1 in the pathogenesis of obesity-induced NAFLD, we generated recombinant adenoviruses that express the NH2-terminal region of mouse XBP-1 (amino acids 1–188), which lacks the transactivation domain and acts in a dominant negative manner to suppress the transcription activity of endogenous XBP-1.33 In ApoE−/−/TLR4-WT mice infused with the recombinant adenoviruses, the ectopic expression of DN XBP-1 in the liver was detectable on day 2 and was still present on day 28 after the initial injection (figure 8A). Compared with ApoE−/−/TLR4-WT mice infused with recombinant adenoviruses expressing luciferase (as a control), ectopic expression of DN XBP-1 resulted in a marked reduction in HFHC diet-induced elevation of histological NASH CRN scores, serum ALT levels, hepatic proinflammatory cytokine production and NF-κB activation (figure 8B–E), as well as a significant decrease in hepatic macrophage infiltration (supplementary figure 5A,B). However, adenovirus-mediated dominant negative suppression of XBP-1 exerted no obvious effect on HFHC diet-induced hepatic lipid accumulation (supplementary figure 5C) and MDA production (figure 8F). These findings suggest that inhibition of XBP-1 is sufficient to block the progression of steatosis to liver inflammation and injury in HFHC diet-fed ApoE−/−/TLR4-WT mice.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of adenovirus-mediated expression of dominant negative (DN) XBP-1 on high-fat, high-cholesterol (HFHC) diet-induced steatosis, malondialdehyde (MDA) production, inflammation and liver injury in ApoE−/−/TLR4-WT mice. TLR4, Toll-like receptor 4; WT, wild-type. ApoE−/−/TLR4-WT mice were fed with standard chow (SC) or an HFHC diet for 8 weeks, followed by tail-vein injection with recombinant adenovirus expressing luciferase (luc) or FLAG-tagged DN XBP-1 (2×108 p.f.u/mouse). (A) Hepatic protein expression of DN XBP-1 was determined by immunoblotting analysis at various days after injection with the recombinant adenoviruses as specified. (B) Representative haematoxylin and eosin-stained liver sections with original magnification at ×400 (upper right) and histological non-alcoholic steatohepatitis (NASH) scores determined according to the NASH Clinical Research Network (CRN) scoring system (lower left). (C) Liver injury was evaluated by measuring serum alanine aminotransferase (ALT) activities. (D) Hepatic mRNA levels of tumour necrosis factor α (TNFα) and monocyte chemoattractant protein-1 (MCP-1). (E) IκB phosphorylation and (F) MDA, were measured at 28 days after injection with recombinant adenoviruses. n=5–8. *p<0.05, **p<0.01. This figure is produced in colour in the online journal—please visit the website to view the colour figure.
Discussion
The lack of suitable animal models that accurately recapitulate the full spectrum of liver pathology of NASH in an appropriate metabolic context is a major obstacle in delineating the mechanisms of this disease.2 A number of genetic and dietary obese models of NAFLD are available, but few show progression to the inflammatory condition of NASH. Although mice fed a methionine-/choline-deficient diet develop the typical symptoms of steatohepatitis, the metabolic profiles of this model are opposite to those in human NASH.2 17 In this study, we demonstrated that ApoE−/−/TLR4-WT mice receiving an HFHC diet for 12 weeks display a liver phenotype strikingly similar to human NASH, including hepatic steatosis, oxidative stress, increased inflammatory infiltration, augmented production of proinflammatory cytokines and raised serum ALT levels. Furthermore, these liver symptoms have developed in a context resembling the metabolic syndrome in humans, including obesity, hyperinsulinaemia, glucose intolerance, dyslipidaemia and atherosclerosis. Therefore, HFHC diet-fed ApoE−/−/TLR4-WT mice can be used as a model to study the progression of simple steatosis to NASH in the context of obesity and metabolic syndrome.
TLR4 and its downstream adaptors are expressed in several types of liver cells, which are constantly confronted with gut-derived LPS.6 7 However, healthy livers do not show signs of inflammation, perhaps owing to the relatively low expression of TLR4 and its downstream adaptors. Mutant mice with non-functional TLR4 are resistant to developing several chronic liver inflammatory diseases.11 13 On the other hand, the precise pathophysiological roles of TLR4 in obesity-related NASH remain poorly defined. This study provides evidence suggesting that activated TLR4 in hepatic macrophages is a key mediator of steatohepatitis in HFHC diet-fed obese mice. This conclusion is supported by our observation that TLR4 expression is selectively elevated in Kupffer cells of ApoE−/−/TLR4-WT mice upon feeding with HFHC diet. Conversely, ApoE−/−/TLR4mut mice, which lack functional TLR4, are resistant to developing HFHC diet-induced steatohepatitis and liver injury, whereas the susceptibility of ApoE−/−/TLR4mut mice to HFHC diet-induced NASH is restored by repopulation of hepatic macrophages with BM cells derived from ApoE−/−/TLR4-WT mice. As Kupffer cells are the major source of production of proinflammatory cytokines in NASH, elevated TLR4 expression in these cells may explain why obese mice are highly susceptible to LPS and develop NASH after a low dose of LPS.34
Upon activation by its ligands, TLR4 triggers NF-κB activation and proinflammatory cytokine production by recruiting the common TLR adaptor myeloid differentiation factor 88 (MyD88).7 However, one study has demonstrated that TLR4-induced liver inflammation and injury in alcoholic liver disease is independent of MyD88,11 suggesting that an alternative mechanism(s) is involved in TLR4-mediated proinflammatory responses in the liver. In this study, we have provided several lines of evidence demonstrating that XBP-1, an important transcription factor that is activated by unconventional mRNA splicing in response to ER stress,35 as a downstream effector of TLR4 mediating HFHC diet-induced liver inflammation and damage in mice. First, an HFHC diet induces the activation of XBP-1 in livers of ApoE−/−/TLR-WT mice, but this effect is abrogated in ApoE−/−/TLR4mut mice. Second, LPS-induced activation of NF-κB and production of proinflammatory cytokines are markedly attenuated by siRNA-mediated knockdown expression of XBP-1. Third, HFHC diet-induced liver inflammation and injury are decreased by adenovirus-mediated expression of a dominant negative XBP-1. In line with our in vivo findings, the activation of XBP-1 by TLRs has also been seen in mouse J774 macrophages.20 XBP-1 is required for optimal and sustained production of proinflammatory cytokines induced by TLR activation, through its direct interaction with the promoter regions of several inflammatory genes (such as TNFα and interleukin 6).20
Besides its role in the UPR, XBP-1 is also an important regulator of adipogenesis and lipogenesis.36 37 In liver, XBP-1 directly controls the induction of critical genes involved in fatty acid synthesis. Selective deletion of XBP-1 in hepatocytes results in a profound reduction in circulating cholesterol and triglycerides in mice.36 Notably, this phenotype is not associated with hepatic steatosis. Likewise, this study demonstrates that adenovirus-mediated expression of dominant negative XBP-1 in the liver has no obvious effect on hepatic lipid accumulation induced by an HFHC diet, but leads to a marked reduction in NF-κB activation, cytokine production and liver injury, suggesting that XBP-1 is mainly involved in the transition of simple steatosis to inflammation.
ER stress, characterised by the activation of three major UPR pathways, has been seen in almost all types of chronic liver diseases in both rodents and humans.38 Unresolved ER stress causes liver damage by increasing lipid synthesis, inducing apoptosis and triggering inflammation.35 38 This study shows that all the three branches of the UPR pathways, including IRE1α/XBP-1, PERK and ATF6 are activated in HFHC diet-induced fatty liver. However, TLR4 inactivation results in differential changes in these UPR pathways, with the activation of the IRE1α/XBP-1 branch being selectively suppressed. Consistent with our observation in HFHC diet-induced obese mice, the differential effects of TLR activation on the three branches of the UPR pathways have been reported in several types of cells as well as in mice treated with a low dose of LPS.20 39 Taken in conjunction, these findings support the notion that the three branches of the UPR pathway can be differentially regulated and also suggest that the role of XBP-1 in mediating TLR4-induced inflammation is unrelated to its function in the UPR.
Besides ER stress, oxidative stress is another critical player in the pathogenesis of chronic liver diseases. In obesity-associated fatty liver, excessive ROS and lipid peroxidation products contribute to the transition of steatosis to steatohepatitis and fibrosis, by inducing inflammation and cell injury.10 Likewise, this study demonstrates that TLR4-induced liver inflammation is mediated in part by augmented ROS production. This conclusion is supported by our findings that HFHC diet-induced elevation in TLR4 protein expression is accompanied by increased lipid peroxidation products in liver from ApoE−/−/TLR4-WT mice, whereas such changes are abrogated in ApoE−/−/TLR4mut mice. In line with our findings, several previous studies have linked the TLR4 signalling with oxidative stress, possibly by enhancing the activity of NADPH oxidase.40 41 Pharmacological inhibition of NADPH oxidase or genetic deletion of Nox (a subunit of NADPH oxidase) protects mice from LPS-induced lethality as well as downregulating the production of inflammatory cytokines.41 Conversely, TLR4 signalling is also required for ROS-induced inflammation and injury, suggesting that a positive feedback regulation between TLR4 and ROS is critical for initiation and amplification of inflammation.10 This notion is further supported by our finding that LPS-induced inflammation in primary rat Kupffer cells is abrogated by either antioxidants or by genetic inhibition of TLR4. Furthermore, our study demonstrates that TLR4 and ROS signalling converge at XBP-1, which in turn potentiates inflammation.
In summary, this study supports the role of TLR4 in Kupffer cells as a central mediator in HFHC diet-induced steatohepatitis and liver damage, partly by inducing ROS-dependent activation of XBP-1 (supplementary figure 6). These findings provide a unified mechanism to explain how multiple ‘hits’ (TLR4 activation, ER stress and oxidative stress) act in concert in promoting the progression of benign steatosis to steatohepatitis in mice. In light of the obligatory role of XBP-1 in TLR4-induced liver inflammation and injury, therapeutic interventions that inhibit XBP-1 activation may represent a promising strategy for treating NAFLD and NASH.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
DY and FYLL contributed equally to this work.
Funding This work is supported by Collaborative Research Fund (HKU5/CRF/08 and HKU4/CRF10) from the Research Grant Council of Hong Kong, Seeding fund for basic research from the University of Hong Kong (to AX), and the National Basic Research Program of China (2011CB504004).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.