Article Text

Abstract
Background and aims: We tested the hypothesis that the actual or predicted consequences of mutations in the cystic fibrosis transmembrane regulator gene correlate with the pancreatic phenotype and with measures of quantitative exocrine pancreatic function.
Methods: We assessed 742 patients with cystic fibrosis for whom genotype and clinical data were available. At diagnosis, 610 were pancreatic insufficient, 110 were pancreatic sufficient, and 22 pancreatic sufficient patients progressed to pancreatic insufficiency after diagnosis.
Results: We identified mutations on both alleles in 633 patients (85.3%), on one allele in 95 (12.8%), and on neither allele in 14 (1.9%). Seventy six different mutations were identified. The most common mutation was ΔF508 (71.3%) followed by G551D (2.9%), G542X (2.3%), 621+1G→T (1.2%), and W1282X (1.2%). Patients were categorized into five classes according to the predicted functional consequences of each mutation. Over 95% of patients with severe class I, II, and III mutations were pancreatic insufficient or progressed to pancreatic insufficiency. In contrast, patients with mild class IV and V mutations were consistently pancreatic sufficient. In all but four cases each genotype correlated exclusively with the pancreatic phenotype. Quantitative data of acinar and ductular secretion were available in 93 patients. Patients with mutations belonging to classes I, II, and III had greatly reduced acinar and ductular function compared with those with class IV or V mutations.
Conclusion: The predicted or known functional consequences of specific mutant alleles correlate with the severity of pancreatic disease in cystic fibrosis.
- cystic fibrosis
- cystic fibrosis transmembrane regulator
- pancreatic insufficiency
- pancreas
- CF, cystic fibrosis
- CFTR, cystic fibrosis transmembrane regulator
- PI, pancreatic insufficiency
- PS, pancreatic sufficiency
Statistics from Altmetric.com
- CF, cystic fibrosis
- CFTR, cystic fibrosis transmembrane regulator
- PI, pancreatic insufficiency
- PS, pancreatic sufficiency
Cystic fibrosis (CF) affects multiple organs, including the pancreas, intestine, sweat gland, and the respiratory tract. Identification of the cystic fibrosis gene1–3 led to the elucidation of its protein product, the cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP regulated chloride channel.4 Mutations in the CFTR gene result in absent or defective CFTR chloride channel function at the apical surface of epithelial cells. The most common disease causing mutation, which accounts for 66–70% of CF chromosomes worldwide, gives rise to a deletion of a single amino acid, phenylalanine, at position 508 (ΔF508) of the CFTR product.2 More than 1000 additional mutations (http//www.genet.sickkids.on.ca/cftr/mutations.html) or putative mutations in the CFTR gene have been identified, all of which are uncommon or rare.
Infants with CF have progressive pancreatic damage leading to signs and symptoms of maldigestion due to pancreatic insufficiency (PI). However, 10–15% of patients retain adequate pancreatic reserve to permit normal digestion without enzyme therapy.5,6 In general, those with the “pancreatic sufficient” (PS) phenotype have less severe disease manifestations.6 We have previously shown that there is a strong clinical association between phenotype and genotype in the exocrine pancreas.7,8 Initial investigations into the phenotype-genotype relationships in CF were made by studying the clinical phenotype and then making an assumption regarding the genotype. For example, R117H was noted to be present in PS patients and therefore an assumption was made that this was a mild mutation.9 From these studies it was determined that most individuals with PI have two severe CFTR mutations while those with the PS phenotype have one or two mild mutations. However, this correlation was limited as it was not based on the molecular mechanisms of the specific mutations. In this study, we attempt to overcome this limitation by classifying the CFTR mutations based on their functional consequences, and then evaluating the relationship with the pancreatic phenotype.
CFTR gene mutations can be classified according to their predicted effects on CFTR mediated anion secretion.9–11 In a five class system, mutations belonging to classes I, II, and III are predicted to have severe functional consequences on CFTR function via different molecular mechanisms. Mutations belonging to classes IV and V on the other hand are expected to confer some residual CFTR mediated channel function and to have milder consequences.
It is our hypothesis that the molecular consequences of CFTR gene mutations correlate with the exocrine pancreatic phenotype in patients with CF. The purpose of this analysis was to validate previous studies of the phenotype-genotype relationship by starting from the basic molecular mechanisms and then evaluating the link with the clinical phenotype. In this report, we present unique data describing the relationship between the predicted consequences of mutations of the CFTR gene and quantitative exocrine pancreatic disease in patients with CF.
METHODS
Patient ascertainment
We assessed all patients with CF attending the Toronto clinics who had undergone genotype analysis. The diagnosis of CF was based on the 1998 consensus conference criteria.12 Each patient’s clinical data and genotype had been recorded in a computerised database. Each patient’s pancreatic status (PS or PI) at diagnosis and subsequent changes in pancreatic status (PS→PI) were defined. Patients who had undergone pancreatic stimulation testing were selected for further analysis of the relationship between exocrine pancreatic function and genotype. The study was approved by the institutional ethics review board.
Assessment of exocrine pancreatic function
Pancreatic function was assessed by one or more of the following methods.
Faecal fat
Three to five day pooled stool collections were stored at 4°C, and faecal fat content was determined by the method of van de Kamer and Weyers13 or by the method of Jeejeebhoy and Kozak.14 Faecal fat losses, expressed as a percentage of mean daily fat intake, were calculated by weighing food intake and by reference to standard food content tables. PI was defined as faecal fat losses exceeding 7% of measured fat intake in patients older than six months, or exceeding 15% in infants younger than six months of age.15
Serum cationic trypsinogen
This radioimmunoassay test reliably distinguishes patients with PS from those with PI after seven years of age, as previously described.16 PS patients with CF have shown widely fluctuating values within or above the normal range. Infants with PI have high serum trypsinogen levels which decline to low or undetectable levels by seven years of age. Patients who are PS at diagnosis but progress to PI (PS→PI) show a progressive but delayed decline in serum trypsinogen levels.17 Patients with PS were tested every 6–12 months. If serum trypsinogen values were declining, or if clinical symptoms suggested pancreatic failure, confirmatory studies (fat balance and/or pancreatic stimulation tests) were performed.
Quantitative pancreatic stimulation test
Patients were assessed by a marker perfusion technique.18 Enzymes (colipase, total lipase, and trypsin),18 and fluid19 and electrolyte (Cl− Na+, HCO3−)20 output were expressed per kg body weight/hour after correcting for the fractional recovery of infused marker. Patients were defined as PI if colipase output was less than 120 u/kg body weight/hour or if trypsin output was below 50 u/kg body weight/hour. If several tests were performed, the results of the most recent test were used.
Genotype analysis
Genotype analysis was performed using total human genomic DNA extracted from peripheral blood cells by standard methods. Each exon of the CFTR gene was amplified by the polymerase chain reaction and screened by SSCP and MDE gel analyses.21 Direct sequencing analysis was used to characterise the DNA sequence of a suspected mutation. Based on retrospective data, this method is capable of detecting more than 95% of known CFTR gene mutations. This detection rate does not include patients with rare unknown mutations or mutations that may lie outside the boundaries of the CFTR gene (that is, in regulatory elements).
Genotype classification
Genotypes were classified according to their known or predicted functional consequences.9–11 With the exception of patients who were homozygous for ΔF508, most patients were compound heterozygotes with mutations belonging to more than one class. We used the functional class of the second mutation on a ΔF508 background. This approach was used to evaluate the effect of the second mutation in patients carrying one ΔF508 mutation. For example, a patient carrying a class I mutation (for example, G542X) in combination with ΔF508 (class II) was classified as class I although the genotype is in fact a compound heterozygote of class I/II. As class IV or V mutations have a dominant effect, patients with these mutations were classified according to the presumed consequences of the dominant mutation.
Statistical analysis
Patient characteristics are presented as mean (SD) and groups were compared using the Student’s t test; χ2 tests were used to compare proportions. Results of the quantitative pancreatic stimulation test produced several measurements with skewed distributions and unequal variance estimates in the subgroups to be compared. Therefore, the Kruskal-Wallis rank sum test22 was used to test subgroup differences in pancreatic secretion measures.
RESULTS
Patients
We assessed 742 patients with a confirmed clinical diagnosis of CF who had been followed between 1990 and 1997 (table 1). At diagnosis, 610 (82.2%) were PI and 110 (14.8%) have remained PS. Twenty two patients, who were PS at diagnosis, became PI during follow up (PS→PI). As previously observed,6,7 patients in the PS group were diagnosed at a later age, had lower mean sweat chloride values, better pulmonary function, better nutritional status, and a lower mortality than those with the PI phenotype. There was no significant difference in sweat chloride, forced expiratory volume in one second, weight for height, or age of diagnosis between the PI and PS→PI groups.
Patient demographics
Genotype analysis
CFTR gene mutations were identified on both alleles in 633 patients (85.3%) and on one allele in only 95 (12.8%) patients. Among the group with only one identified mutation, 40 were PS at diagnosis. No CFTR gene mutations were identified on either allele in 14 (1.9%) patients, of whom seven were PS at diagnosis. Seventy six different CFTR gene mutations were identified among the 1324 chromosomes in 662 families (table 2). The most common mutations were: ΔF508 (in 943 chromosomes, 71.2%), G551D (39, 2.9%), G542X (31, 2.3%), 621+1G→T, W1282X (16, 1.2%), and R117H (11, 0.8%). Less common mutations included G85E and 5T (n=5 chromosomes), A455E and R1162X (n=4 chromosomes), R347, Y1092X, R334W, and V520F (n=3 chromosomes). In 46 CF chromosomes a mutation was found that was absent in other tested families, and in 116 CF chromosomes no mutation was detected.
Genotype classification according to the functional consequences of CFTR gene mutations
Analysis of 633 patients with two identified CFTR gene mutations confirmed our previously documented association between pancreatic phenotype and severe and mild genotypes,8,10 and provided further information concerning the phenotypic effects of more recently identified gene mutations. Mutations were classified as mild or severe, as previously described.7 With very few exceptions, two severe mutations conferred PI and one mild mutant allele conferred PS in a dominant manner. Among the 569 patients carrying two severe mutations, 546 (96.0%) were PI, while 61/62 (98%) with at least one mild mutation were PS. A subset of patients with severe mutations on both alleles were PS at diagnosis but progressed to PI. Only four individuals (0.7%) with two severe mutations have remained PS. Pancreatic stimulation testing of one patient (ΔF508/ΔF508) revealed markedly reduced pancreatic acinar function, close to the threshold for developing PI.18 The three remaining patients were not tested.
Functional consequences of CFTR gene mutations and pancreatic phenotype
We were able to classify 607 of the 633 patients according to the predicted consequences of their CFTR gene mutations (table 2). Patients who did not carry ΔF508 but who were compound heterozygotes for mutations in different classes were excluded. Ninety nine per cent (552/557) of patients with both mutations homozygous or compound heterozygous for classes I, II, or III were PI at diagnosis (528), or progressed to PI after diagnosis.19 Only five individuals with class I, II, or III mutations have remained PS since diagnosis. This includes one patient who carries the missense mutation G85E which appears to have an indeterminate effect on pancreatic phenotype as it has been observed in patients with PS and PI phenotypes.23 All but one of 49 individuals carrying at least one mutation belonging to classes IV or V have remained PS. The exception, a patient with the class IV mutation I1234V, developed PI at the age of 25 years, after symptoms of chronic pancreatitis lasting 10 years.
Genotypes and quantitative measures of pancreatic function
Pancreatic function testing was performed in 95 patients (34 female), including 46 who have remained PS and 49 who were either PI at diagnosis or developed PI after diagnosis. Table 3 shows pancreatic acinar and ductular function data according to the functional classification system for CFTR gene mutations. All patients who were homozygous or compound heterozygous for mutations belonging to classes I, II, or III had severely impaired pancreatic acinar and ductular function. When expressed as a percentage of mean output reference values (fig 1) there was a marked deficit of acinar capacity in patients with class I, II, or III mutations compared with those with class IV or V mutations (p<0.0001, Kruskal- Wallis, χ2, 4 df). Among patients with class IV and V mutations, acinar function spanned a wide range of enzyme output from 18% of mean control values to values well within the reference range. Similarly, ductular function (fluid, bicarbonate, and chloride secretion) was significantly lower among patients homozygous or compound heterozygous for class I, II, or III mutations than that observed in patients with mutations belonging to class IV or V (p<0.01 for H2O; p<0.001 for HCO3− and Cl−). Patients in classes I, II, and III who were PS→PI were indistinguishable from those who were PI at diagnosis. Acinar output in the PS→PI group ranged from 0% to 14.3% of control values.
Pancreatic ductular and acinar secretion classified according to the functional consequences of CFTR gene mutations
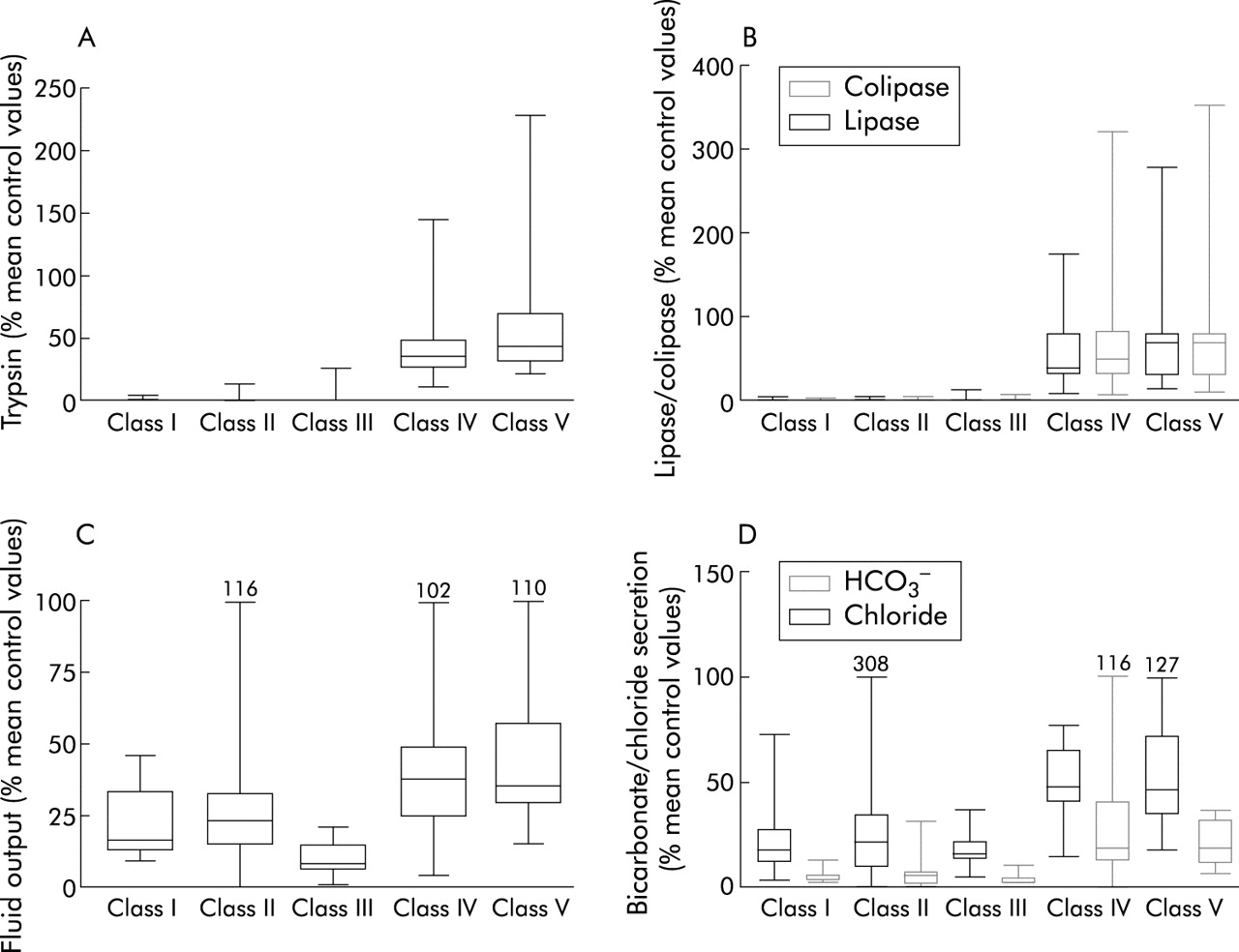
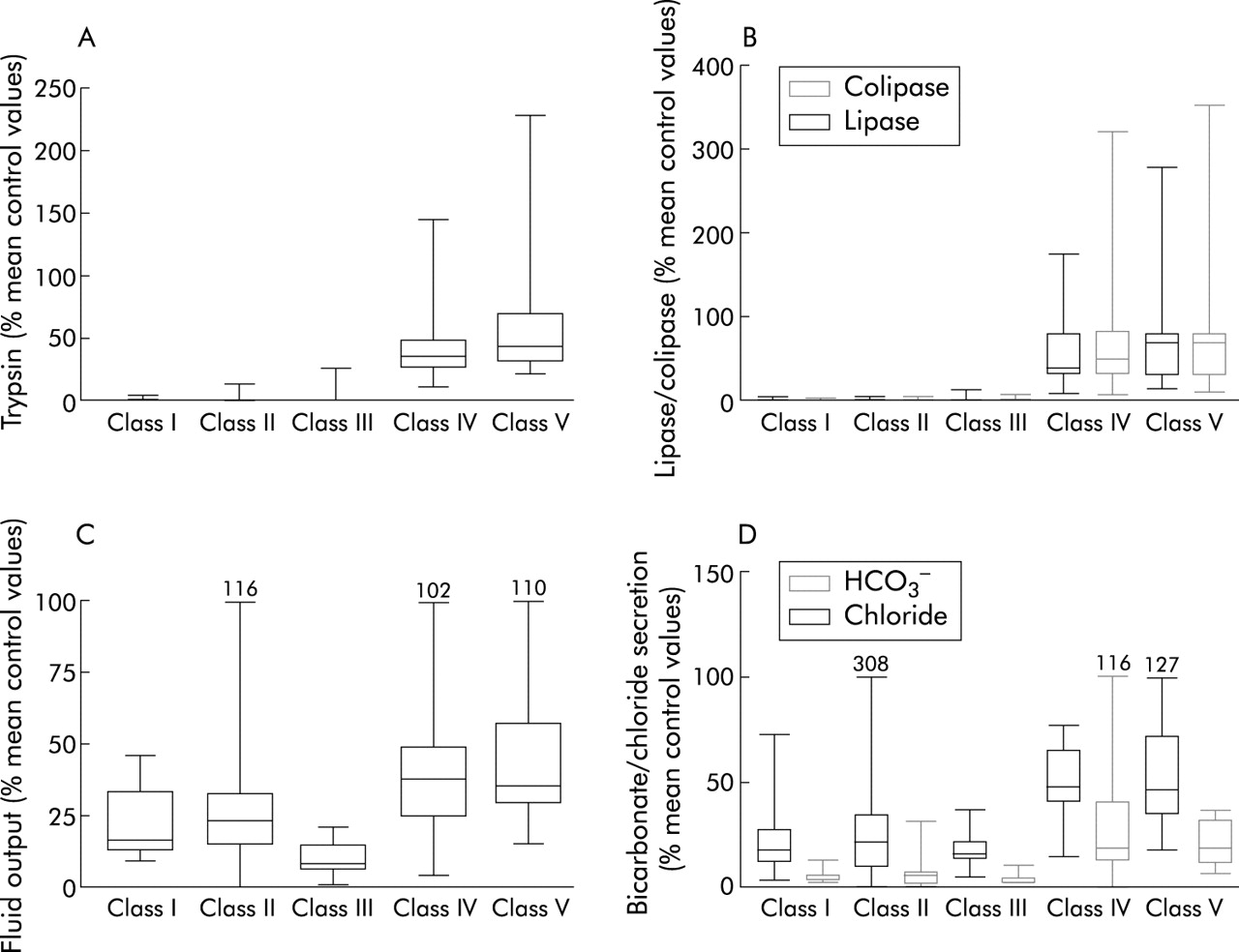{kind=link}
Quantitative pancreatic acinar and ductular secretion following intravenous cholecystokinin/secretin infusion in 70 patients, expressed as a percentage of mean control values. (A) Trypsin; (B) lipase and colipase; (C) fluid output; and (D) bicarbonate (HCO3−) and chloride secretion are shown. Box margins represent the 25th and 75th percentiles, with the horizontal bar representing the median. Whiskers indicate the range of data. Comparison of classes I, II, and III with classes IV and V showed significant differences: p=0.01 (fluid), p=0.001 (bicarbonate and chloride), p<0.0001 (trypsin, colipase and lipase) by Kruskal-Wallis, χ2, with 4 df.
DISCUSSION
Attempts to elucidate the relation between genotype and the various phenotypic manifestations of CF have been hampered by the large number of CFTR gene mutations. In this study, the frequency of the most common disease causing mutation (ΔF508) was very similar to the frequency of approximately 70% of all CFTR mutant alleles reported worldwide. The four next most common mutations reported in this study (G551D, G542X, 621+1G→T, and W1282X) each accounted for between 1% to 3% of the mutations analysed. Twenty five additional mutations, including the 5 thymidine variant in intron 8,24 were identified in more than two families. Thus 31 CFTR gene mutations or variants which were identified in more than one family accounted for 87.8% of all mutant alleles. Forty five additional mutations, or putative mutations, which were identified in single patients, accounted for only 3.5% of mutant alleles. Despite our attempts to screen all exons of the CFTR gene, 8.8% of mutant alleles remained unidentified. These results are consistent with the recently published CFTR mutation frequencies in the French population.25
Recognising the frequency of uncommon or rare mutations in CF, we evaluated the relation between genotype and pancreatic phenotype by classifying individual patients’ CFTR gene mutations. In this analysis, no assumptions were made about the genotype based on the prior knowledge of the pancreatic phenotype, as genotypes were classified purely based on their known or predicted consequences on CFTR mediated anion secretion (fig 1). CFTR gene mutations belonging to classes I, II, or III have varying effects on gene transcription, mRNA translation, or intracellular trafficking of the nascent protein. In functional terms, however, they would all be expected to confer complete loss of cAMP regulated chloride channel function. The results of the present study conclusively indicate that homozygosity or compound heterozygosity for mutations in classes I, II, and/or III is strongly associated with severe pancreatic disease as the vast majority of these patients were PI at diagnosis or progressed to PI after the diagnosis of CF was established. By contrast, class IV and V mutations are predicted to be less severe in terms of their effects on cAMP regulated chloride channel properties. In our study, all patients carrying at least one class IV or class V mutation were PS at diagnosis, as measured by pancreatic function testing, and all but one have remained PS. The single exception appeared to develop PI due to progressive pancreatic damage from chronic pancreatitis. In support of these results, data from the European Epidemiology Registry of Cystic Fibrosis showed that patients with class IV and V mutations were less likely to be taking pancreatic enzyme supplements. However, pancreatic function status was not objectively defined.26
A subset of patients who had undergone pancreatic stimulation testing were also evaluated according to the predicted consequences of their CFTR gene mutations. Not surprisingly, patients who were homozygous or compound heterozygous for mutations belonging to classes I, II, or III had severely compromised acinar capacity (enzyme secretion of trypsin, total lipase, and colipase). Among the PS→PI patients who were PS when tested (n=14), enzyme secretion was also severely compromised (approximating 0–14.3% of mean control values). The majority of these patients, who carried severe CFTR gene mutation on both alleles, developed PI within two years of life. In contrast, patients in this study with at least one mild class IV or V mutation had significantly better pancreatic acinar reserve, which in the majority has been maintained for decades. Trypsin secretion in this group ranged from 11% of mean control values to values within our adult reference range.18
Pancreatic ductular secretion of anions (chloride and bicarbonate) and fluid was compromised in patients with CFTR gene mutations belonging to all five functional classes. This was expected because CFTR is normally expressed at high levels in the intralobular and proximal ductal epithelia, and at low levels in acinar cells.27,28 CFTR in the ductal epithelium facilitates the secretion of an alkaline rich fluid which maintain the solubility of secreted enzymes. Complete loss of CFTR function results in reduced intraluminal pH and a low secretory volume.29 Scheele and colleagues29 have suggested that reduced pH within the acinar lumen inhibits acinar endocytosis of secretory granule proteins and reduces the solubility of secreted luminal proteins within the acinar lumen. Complete loss of CFTR function, due to mutations on both alleles (classes I, II, or III) appears to induce rapid pancreatic atrophy through obstruction of secreted proteins within the lumina of acini and small ducts. Postmortem studies of the CF pancreas from preterm infants and neonates clearly demonstrate that this process begins in utero30 and explains the rapid progression of pancreatic failure in early infancy. Patients carrying at least one mutant allele belonging to classes IV or V also had evidence of ductular dysfunction. Chloride, bicarbonate, and fluid secretion was significantly less than that observed in adult controls even in those with normal acinar reserve. This observation supports our previous hypothesis that a major component of anion and fluid secretion is directly attributable to functional CFTR in pancreatic ductal epithelia.19,20 Obviously, mild class IV or V CFTR gene mutations, which result in some residual ductular function, appear to protect the exocrine pancreas from complete destruction at an early age.
There are several possible explanations for the heterogeneity of acinar reserve and ductular dysfunction among patients carrying class IV and V mutations. Specific mutant alleles belonging to classes IV and V may exhibit varying degrees of chloride channel dysfunction, which in turn may influence disease progression. Secondly, patients carrying at least one class IV or V mutation may develop progressive pancreatic damage as a consequence of chronic pancreatitis. While the prevalence of acute recurrent pancreatitis or chronic pancreatitis is low in patients with CF diagnosed on clinical grounds,31 those with the PS phenotype appear to carry a high risk of this complication.31
The limitations of this classification system are acknowledged. Classification is not possible if a patient carries a “putative” mutation or if the molecular properties of a disease causing mutation are not understood. Furthermore, despite extensive efforts to identify all CFTR gene mutations, one or two mutant alleles remain unidentified in 17% of patients. A small number of mutations (for example, G85E) are considered to be indeterminate due to their inconsistent effect on pancreatic phenotype. Finally, it is more difficult to predict the functional consequences in compound heterozygotes with mutant alleles belonging to two different classes. Despite these limitations, we have overcome previous assumptions involved in analysing the relationship between genotype and phenotype in CF by starting our analysis with the predicted or known molecular consequences of the specific CFTR mutations. We have demonstrated a strong relation between the predicted functional effects of each genotype and pancreatic phenotype in this patient cohort. Nevertheless, there were a few exceptions. For example, two siblings with a class I mutation (875+1G>C) had a PS phenotype. The precise reason for this is unclear as 875+1G>C adversely affects the highly conserved splice donor site sequence. One possibility is that alternative splicing nearby may preserve the effect of this mutant allele.
In conclusion, we have demonstrated, using analysis of the molecular consequences of CFTR mutations, that the CF genotype strongly correlates with the pancreatic phenotype. Furthermore, we provide unique data indicating that CFTR gene mutations correlate with quantitative measures of pancreatic acinar and ductular function.
Acknowledgments
Supported by grants in aid from the Canadian Cystic Fibrosis Foundation and the National Institute of Health (NIDDK-DK 49096). Dr Ahmed is the recipient of a research fellowship from the Medical Research Council (Canada)