Article Text

Abstract
Traditionally, Crohn’s disease has been associated with a Th1 cytokine profile, while Th2 cytokines are modulators of ulcerative colitis. This concept has been challenged by the description of tolerising regulatory T cells (Treg) and by proinflammatory Th17 cells, a novel T cell population characterised by the master transcription factor RORγt, the surface markers IL23R and CCR6, and by production of the proinflammatory cytokines IL17A, IL17F, IL21, IL22 and IL26, and the chemokine CCL20. Th17 cells differentiate under the influence of IL1β, IL6, IL21 and IL23. Recent studies indicate that TGFβ is essential not only for the development of murine Th17 cells but also for differentiation of human Th17 cells. TGFβ reciprocally regulates the differentiation of inflammatory Th17 cells and suppressive Treg subsets, with the concomitant presence of proinflammatory cytokines favouring Th17 cell differentiation. Several studies demonstrated an important role of Th17 cells in intestinal inflammation, particularly in Crohn’s disease. Genome-wide association studies indicate that IL23R and five additional genes involved in Th17 differentiation (IL12B, JAK2, STAT3, CCR6 and TNFSF15) are associated with susceptibility to Crohn’s disease and partly also to ulcerative colitis. Taken together, both Th1 and Th17 cells are important mediators of inflammation in Crohn’s disease, although activities previously ascribed to IL12 may be mediated by IL23. Anti-IL12/IL23p40 antibody therapy, which targets both Th1 and Th17 cells, is effective in Crohn’s disease. However, the complex relationship between Th1 and Th17 cells has not been completely analysed. This will be of great importance to delineate the specific contributions of these cells to Crohn’s disease and other autoimmune diseases.
Statistics from Altmetric.com
Crohn’s disease and ulcerative colitis are the two main disease entities of inflammatory bowel diseases (IBDs). Although their exact aetiology is still not completely understood, it has been proposed that their pathogenesis is characterised by an exaggerated immune response in genetically susceptible individuals.1 Two decades ago, two T helper (Th) cell subsets were described.2 For many years it has been assumed that Crohn’s disease is mainly mediated by Th1 cells, while ulcerative colitis is a Th2-like type of inflammation. This has been supported by increased levels of Th1 cytokines such as interferon γ (IFNγ) and interleukin 12 (IL12) in Crohn’s disease and an increased expression of certain Th2 cytokines such as IL13 in ulcerative colitis.3–5
Th1 cells produce IFNγ and their primary role is the protection against intracellular microbes (fig 1). Th2 cells produce IL4, IL5 and IL13 and they are involved in allergic disorders and protection against extracellular pathogens including gastrointestinal nematodes (fig 1). Th1 and Th2 cell development are under the control of certain transcription factors including T box expressed in T cells (T-bet) and signal transducer and activator of transcription (STAT) 4 for Th1 cells and GATA-binding protein (GATA)-3 and STAT6 for Th2 cells6 (fig 1). Th1 differentiation is mainly driven by IL12 and IFNγ, while IL4 (in the absence of IL12) drives Th2 differentiation.7

Development of T helper (Th)1, Th2, Th17 and iTreg cells from naïve CD4+ T cells. Cytokines inducing the development of Th1, Th2, Th17 and iTreg cells are marked in red. The main effector cytokines of these four cell lines are marked in blue. Cytokines which inhibit the development of Th17 cells are marked in black. Although IL27 and interferon γ (IFNγ) almost completely suppress the generation of Th17 cells by IL6 and transforming growth factor β (TGFβ), these cytokines only partially inhibit the expression of retinoid-related orphan receptor-γt (RORγt) by IL6 and TGFβ.12 *Murine Treg cells but not human Treg cells produce constitutively IL35, which has anti-inflammatory properties and limits the expansion of Th17 cells. GATA, GATA binding protein; ICOS, inducible co-stimulatory molecule; IL, interleukin; RUNX, runt-related transcription factor; FOXP, forkhead box P3; STAT, signal transducer and activator of transcription; T-bet, T box expressed in T cells.
Recently, a number of other T cell populations were discovered, of which regulatory T cells (Treg) and Th17 cells are the most prominent (fig 1). Treg cells are important for the control of immune responses to self-antigens preventing autoimmunity and maintaining self-tolerance. In contrast, IL17-producing Th17 cells were recognised as novel group of T cells which play a major role in autoimmunity. The intestinal immune system has to maintain both a state of tolerance toward intestinal antigens and the ability to combat pathogens. This balance is achieved by a number of mechanisms including reciprocal regulation of proinflammatory, effector CD4+ cells and tolerising, suppressive Treg subsets. In Crohn’s disease, this balance is lost and the effects of proinflammatory T cells outnumber the tolerising, anti-inflammatory effects of Treg cells (fig 2). Several studies indicated that, in addition to Th1 cells, Th17 cells also play a major role in the pathogenesis of Crohn’s disease. The important role of Th17 cells, which express the IL23 receptor (IL23R) on their surface, in the pathogenesis of Crohn’s disease is also supported by recent genome-wide association studies, demonstrating IL23R and other genes involved in the differentiation of Th17 cells as IBD susceptibility genes.8–11 Therefore, the previous concept of the Th1 and Th2 cell paradigm in IBD has to be revised and up-dated.
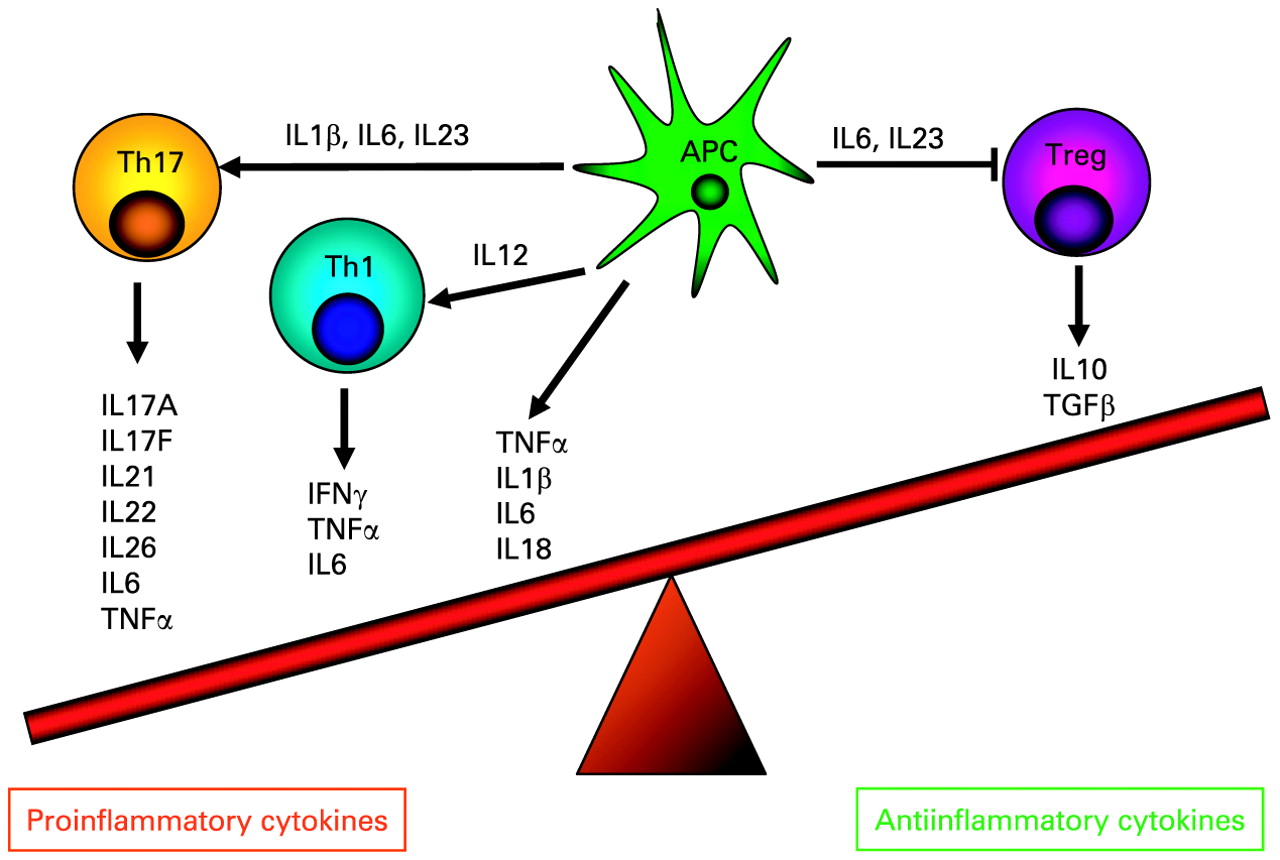
The imbalance of pro- and anti-inflammatory cytokines in Crohn’s disease. In patients with Crohn’s disease, proinflammatory T helper (Th)17 and Th1 cytokines outweigh the effect of anti-inflammatory cytokines secreted by regulatory T cells (Treg). Cytokines released by antigen presenting cells (APCs) following contact with microbial products trigger the differentiation of Th1 and Th17 cells or have a direct proinflammatory effect (eg, via secretion of tumour necrosis factor α (TNFα)). New evidence indicates that interleukin (IL)23 may restrain regulatory T cell activity causing intestinal inflammation.33
THE ROLE OF TH1 CELLS IN THE PATHOGENESIS OF CROHN’S DISEASE
Data gathered before the full characterisation of Th17 cells implicated Th1 cells as the main mediators of the pathogenesis of Crohn’s disease. This was based on a number of observations, including the increased production of IL12 in macrophages in Crohn’s disease.3 13 14 Moreover, nuclear extracts from T cells isolated from inflamed Crohn’s disease lesions contain high levels of activated STAT4 and T-bet, Th1-associated transcription factors indicative of IL12 signalling.15 Furthermore, in the mucosa of patients with Crohn’s disease, there is an enhanced production of IL18, a cytokine involved in perpetuating Th1 cell responses.16 17 In addition, T cells isolated from areas with active Crohn’s disease express increased levels of the IL12Rβ2 chain,18 which is characteristic for Th1 cells, and increased amounts of the Th1 cytokine IFNγ together with decreased levels of the Th2 cytokine IL4 compared to T cells isolated from controls.3 19 IFNγ-secreting lamina propria lymphocytes are abundant in the mucosa of patients with Crohn’s disease. Particularly at onset of Crohn’s disease, mucosal T cells appear to mount a typical Th1 response that resembles an acute infectious process, and is lost with progression to late Crohn’s disease.20 In addition, clinical response was observed in a subcohort of patients with Crohn’s disease treated with an anti-IFNγ antibody.21 A key role for IFNγ has also been shown in a murine transfer colitis model which did not develop after transfer of T cells from IFNγ knockout mice,22 while over-expression of the Th1-related transcription factor STAT4 causes colitis.23
THE ROLE OF IL23 AND TH17 CELLS IN INTESTINAL INFLAMMATION
The most potent Th1-polarising cytokine is IL12, composed of the p40 and p35 subunits. However, its functional role has to be partly re-evaluated given that many studies used antibodies directed against the IL12/IL23p40 subunit, which is also part of the IL23 heterodimer, which is composed of p40 and the unique subunit p19. Studies in autoimmune models traditionally recognised as Th1 disease models revealed that tissue inflammation did not develop in mice deficient in the IL23p19 subunit, while inflammation was seen in IL12p35-deficient mice24 25 suggesting that these models of autoimmunity are linked to Th17 and not primarily Th1 responses. Similar observations were made in models of intestinal inflammation. Hue et al demonstrated that IL23p19 antibodies suppress chronic intestinal inflammation in T cell-deficient Helicobacter hepaticus-infected RAG knockout mice.26 These findings were supported by the results of a transfer colitis model. In that model recipient mice deficient in IL23p19 and IL12/IL23p40 are protected against colitis, while mice deficient for the IL12-specific subunit p35 were not protected against colitis.27 This clearly indicates a more important role for IL23-driven Th17 cytokines than for IL12-driven Th1 cytokines in these mouse models of intestinal inflammation. Similarly, CD40-induced colitis depended on IL23p19 secretion, while IL12p35 secretion controlled wasting disease and serum cytokine production but not mucosal immunopathology.28 In agreement, Yen et al demonstrated that colitis in IL10-deficient mice was dependent on IL23 rather than IL12, further supporting the role of IL23-driven Th17 cytokines in intestinal inflammation.29 Moreover, colitis could be exaggerated by exogenous administration of recombinant IL23,29 while in another murine colitis model an anti-IL23p19 antibody treated colitis.30
However, given that IL23p19-deficient mice are highly susceptible to T cell-mediated colitis induced by a hapten reagent,31 it is assumed that selective neutralisation of IL23 may not be beneficial under certain circumstances. Becker et al revealed that dendritic cells (DCs) from IL23p19-deficient mice produced elevated levels of IL12, and that IL23 downregulates IL12 expression upon TLR ligation.31 Interestingly, in vivo blockade of IL12p40 in IL23p19-deficient mice rescued mice from lethal colitis,31 suggesting cross-regulation of IL12 expression by IL23 as a novel key regulatory pathway during initiation of T cell-dependent colitis.32
Novel data also indicate that IL23 restrains Treg activity to drive T cell-dependent colitis.33 The frequency of naïve T cell-derived forkhead box P3 (FOXP3)+ cells in the colon increased in the absence of IL23, indicating a role for IL23 in controlling Treg induction.33 FOXP3-deficient T cells induced colitis when transferred into recipients lacking IL23p19, showing that IL23 was not essential for intestinal inflammation in the absence of FOXP3.33 These data indicate that over-riding immunosuppressive pathways is an important function of IL23 in the intestine.
The findings of murine models of intestinal inflammation are supported by findings in human IBD. Studies in Crohn’s disease showed increased IL12 and IL23 expression levels.34 35 Therefore, both inhibiting Th1 cell-activating IL12 and Th17-activating IL23 may have therapeutic potential. In agreement with these observations, an anti-IL12/IL23p40 antibody (directed against the common p40 subunit of IL12 and IL23) demonstrated its clinical potential showing a high rate of remission and clinical responses compared to placebo.36 Ustekinumab, a similar antibody directed against the p40 subunit of IL12 and IL23, induced clinical response in patients with moderate-to-severe Crohn’s disease, especially in patients previously given infliximab.37 Currently, there are no clinical data on the efficacy of an antibody specifically neutralising the IL23p19 unit in human intestinal inflammation, but based on its efficacy in murine intestinal inflammation,30 future trials using such an antibody in patients with Crohn’s disease are anticipated. This would also help to clarify whether antagonising the Th1 mediator IL12 or neutralising Th17-specific IL23 is responsible for the treatment success seen with the IL12/23p40 antibody which neutralises both cytokines.
Proinflammatory cytokines such as TNFα, IL6 and IL1β primarily released from macrophages and DCs also play a major role in the perpetuation of the intestinal inflammation in Crohn’s disease resulting in an imbalance of pro- and anti-inflammatory cytokines (fig 2). Th1 cells, as well as Th17 cells, may contribute to the increased expression of TNFα in Crohn’s disease,38 39 suggesting that the strong anti-inflammatory effects of anti-TNF antibodies in Crohn’s disease may also contribute to their ability to limit Th17-mediated inflammation. Interestingly, in patients with Crohn’s disease, one study identified a unique subset of CD14+ macrophages that contributed to IFNγ production rather than IL17 production by lamina propria mononuclear cells (LPMCs) dependent on IL23 and TNFα.40 The same group also found significant IL17 mRNA upregulation in lamina propria CD4+ cells from patients with ulcerative colitis, while IFNγ was increased in Crohn’s disease41 arguing somewhat against the concept that IL23 contributes only to Th17 cytokine production.
HUMAN TH17 AND TH17/TH1 CELLS AND THEIR INVOLVEMENT IN THE IMMUNOPATHOLOGY OF CROHN’S DISEASE
Although many studies analysed the role of Th17 cells in animal models of intestinal inflammation and autoimmunity, there are only a few studies investigating the role of Th17 cells in patients with Crohn’s disease. We recently demonstrated an increased number of T cells expressing retinoid-related orphan receptor-γt (RORγt), the master transcription factor for Th17 cells, in the lamina propria of patients with Crohn’s disease.42 Th17 cells were also isolated from inflamed lesions of patients with Crohn’s disease by Pene et al.43 Similar to Th2 cells, Th1 cells negatively cross-regulate the differentiation of Th17 cells (fig 1), and mice lacking the Th1 transcription factor T-bet are characterised by greater numbers of Th17 cells,44 which might be related to reduced IFNγ production, resulting in less inhibition of Th17 cell development. However, a number of investigators identified a subset of Th17 cells which may co-produce the Th1 cytokine IFNγ.45 46 This is particularly prominent at sites of inflammation such as active Crohn’s disease,45 This may suggest that IFNγ may not always downregulate IL17A production and may contribute to the pathogenetic and proinflammatory functions of Th17 cells, although this hypothesis needs further investigation.
Two independent studies demonstrated Th17 cells in human peripheral blood and in the gut from healthy individuals and patients with Crohn’s disease.45 46 These two studies showed that these cells are characterised by the expression of RORγt, IL23R and CCR6, whereas they lack CXCR3, a chemokine receptor that is characteristic for Th1 cells45 46 (fig 3). The study by Annunziato et al demonstrated IL17A-producing T cells in the gut, including T cell populations which also expressed both IL17A and IFNγ, which they named “Th17/Th1” cells45 (fig 3). Acosta-Rodriguez et al identified Th17 cells to be characterised by CCR6+CCR4+ expression, while CCR6+CXCR3+-expressing Th1 cells also included a subset which produced both IL17A and IFNγ46 (fig 3). Moreover, very recent findings implicate CD161 as a novel surface marker for human Th17 cells and demonstrate the exclusive origin of these cells from a CD161+CD4+ T cell progenitor49 (fig 3). The studies by Annuziato et al45 and by Acosta-Rodriguez et al46 challenge the traditional concept that IFNγ suppresses Th17 and enhances Th1 development (fig 1). This is in line with a very recent study demonstrating that IFNγ triggers APCs to produce IL1 and IL23 and enables them to induce memory Th17 cell expansion.50 Therefore, the interactions between Th1 and Th17 cells and the role of IFNγ on Th17 cells may be more complex than previously assumed and require further analysis to delineate the specific contributions of these cell lines to Crohn’s disease and other autoimmune diseases.
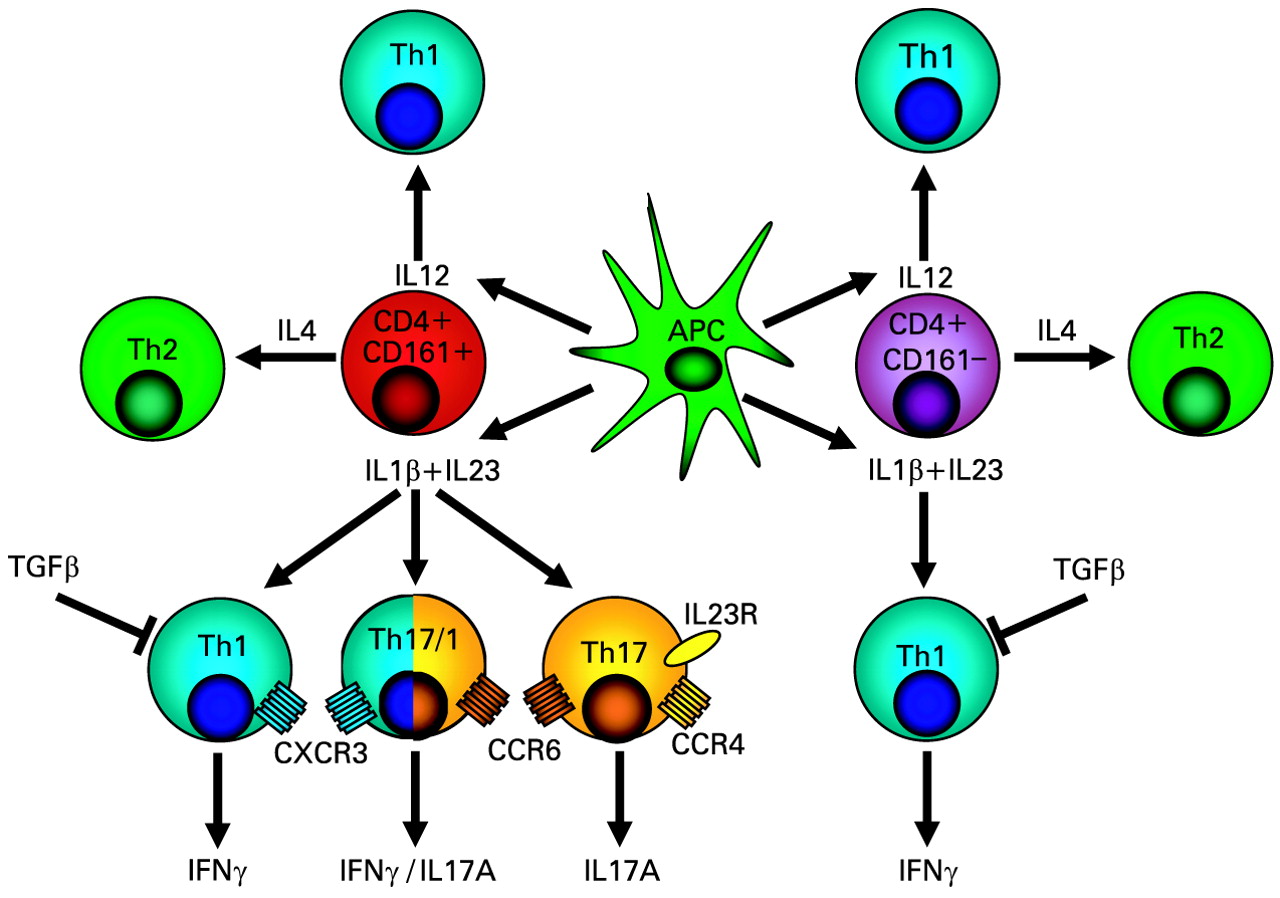
Development of human T helper (Th)1, Th17 and Th17/Th1 cells from a common CD161+CD4+ precursor cell. Human Th17 and Th17/Th1 cells, which show features of both Th17 and Th1 cells, originate only from CD161+CD4+ T cell precursor cells, while Th1 and Th2 cells can originate from both CD161+ and CD161− T cells. Essential for Th17 and Th17/Th1 cell development from CD4+CD161+ precursors is exposure to antigen presenting cell (APC)-released IL23 and IL1β, while IL12 favours the development of Th1 cells and IL4 the development of Th2 cells. Th17 cells are characterised by surface expression of CCR6 and CCR4, while CCR6+CXCR3+-expressing Th1 cells include a subset which produces both IL17A and IFNγ (“Th17/Th1 cells”). Another study demonstrated that human Th17 cells are identified as bearing the CCR2+CCR5−phenotype.47 This schematic drawing is based on a figure by Annunziato et al48 and the results of the study by Acosta-Rodriguez et al.46 IFN, interferon; IL, interleukin; TGF, transforming growth factor.
TH17 CELL DIFFERENTIATION
In contrast to Th1 and Th2 cell differentiation, which depend on their respective effector cytokines (IFNγ and IL4) for differentiation (fig 1), Th17 cells do not require IL17 for their differentiation. Murine Th17 cells originate from naïve CD4+ T cells in the presence of IL6 and TGFβ and their development is then amplified and stabilised by IL21 and IL2351–53 (fig 4). RORγt has been identified as the master transcription factor guiding Th17 differentiation, which also synergises with other transcription factors such as STAT3, RORα, interferon regulatory factor (IRF)4 and runt-related transcription factor (RUNX)1.54–57 IL23 promotes maintenance but not commitment to the Th17 lineage.58

Th17 cell differentiation in mice and humans. (A) Murine Th17 cells originate from naïve CD4+ T cells in the presence of TGFβ (likely secreted by iTregs) and IL6 (secreted by APCs). Their development is then amplified by IL21 and stabilised by IL23. Th17 cells secrete and respond to IL21 in an autocrine manner. IL12, IFNγ, IL2, IL4, IL25, IL27, IL35 and retinoid acid inhibit the development of murine Th17 cells. In contrast, the presence of TGFβ and retinoic acid or IL2 reciprocally favours the development of tolerising, anti-inflammatory iTregs. (B) Human Th17 cells seem to originate in response to the combined activity of IL1β and IL6, or the activity of IL1β or IL23 alone, whereas the combined activity of IL1β and IL23 was not shown to have any additive or synergistic effect. Addition of TGFβ (*) to human naïve or memory CD4+ T cells inhibited the development of human Th17 cells in some earlier studies.59–61 However, very recently, TGFβ in combination with IL1β, IL6 or IL21 was shown to induce the differentiation of human Th17 cells.62 63 This indicates that there are likely no major differences in the differentiation of murine and human Th17 cells. Similar to murine Th17 cells, IL21 and IL23 amplify and stabilise the development of human Th17 cells. IL23R and CCR6 are surface markers of Th17 cells. APC, antigen presenting cell; IFN, interferon; IL, interleukin; TGF, transforming growth factor; Th, T helper.
Initial studies demonstrated differences between the development of murine and human Th17 cells. In particular, it was questioned whether TGFβ, which is essential for the development of murine Th17 cells, is also required for the development of human Th17 cells.59–61 However, very recently, TGFβ in combination with IL1β, IL6 or IL21 was shown to induce the differentiation of human Th17 cells,62 63 suggesting no major differences between the differentiation of mouse and human Th17 cells, although some aspects were investigated in greater detail in murine Th17 cells (fig 4). The discrepancy between the different studies regarding the effect of TGFβ might be related to its capacity to upregulate RORγt and simultaneously to inhibit its ability to induce IL17 expression.62 Inflammatory cytokines relieve this inhibition and increase RORγt-directed IL17 expression.62
In addition to its role in Th17 differentiation, TGFβ induces the Treg-specific transcription factor FOXP364 which is a key regulatory gene for the development of Treg cells.65 Given that addition of IL6 to TGFβ inhibits the generation of Treg cells and induces Th17 cells, a reciprocal relationship between Treg cells and Th17 cells has been proposed.52 In murine T cells, the vitamin A metabolite retinoic acid is a key regulator in TGFβ-induced T cell differentiation.66 While retinoic acid inhibits the Th17 cell-associated transcription factor RORγt in Th17 cell-inducing conditions, it promotes the differentiation of TGFβ-dependent Treg cells66–69 (fig 4). The reciprocal relationship between Treg and Th17 cells is further supported by the fact that IL2, which is a growth factor for Treg cells, inhibits the generation of Th17 cells70 (fig 4). Similarly, the aryl hydrocarbon receptor was identified as a ligand-dependent system to control the generation of Th17 versus iTreg cells.71
THE ROLE OF INTESTINAL MICROBIOTA IN TH17 CELL DIFFERENTIATION
The development of intestinal Th17 cells is also influenced by the gut microbial flora.72 In specific pathogen-free mice, Niess et al demonstrated high numbers of Th17 cells in the lamina propria of the ileum and colon, areas which are typically affected by Crohn’s disease.73 They demonstrated that the microflora is required for the accumulation of cytokine (IL17A, IFNγ, TNFα, IL10)-producing CD4+ T cells in the colonic lamina propria.73 In agreement with this study, analysis of different mouse strains performed in Dan Littman’s laboratory demonstrated that Th17 cell differentiation in the lamina propria of the small intestine requires specific commensal microbiota and is inhibited by treating mice with selective antibiotics.74 The absence of Th17 cell-inducing bacteria was accompanied by an increase in FOXP3+ Tregs in the lamina propria.74 These results suggest that the composition of intestinal microbiota regulates the Th17:Treg balance in the lamina propria and may thus influence intestinal immunity and tolerance which is disturbed in Crohn’s disease.74 While some bacteria favour Th17 cell development, certain commensals and probiotics may limit uncontrolled Th17 cell development. In germ-free mice, Zaph et al demonstrated that the frequency of Th17 cells in the large intestine is significantly elevated in the absence of commensal bacteria.75 Commensal-dependent expression of the IL17 family member IL25 (IL17E) by intestinal epithelial cells limited the expansion of Th17 cells in the intestine by inhibiting expression of macrophage-derived IL2375 (figs 1 and 5). Certain commensals also limited Th17 production in other studies.76 77

Interactions between cytokines released from Th17 cells, APCs and intestinal epithelial cells in Crohn’s disease. After activation of TLRs and NOD2 by bacterial products, APCs secrete IL23, favouring the development of Th17 cells, and IL12, which is essential for the development of Th1 cells. Commensal-dependent expression of IL25 (IL17E) by IECs limits the expansion of Th17 cells in the intestine by inhibiting expression of APC-derived IL23. Th17 cells secrete a number of proinflammatory cytokines for which receptors are expressed on IECs. IECs release in turn an array of proinflammatory cytokines and chemokines such as CXCL8 attracting neutrophils and CCL20, which is chemotactic for Th17 cells and DCs, further exacerbating the intestinal inflammation. Certain Th17 cytokines such as IL22 have also epithelial barrier protective properties such as increasing defensin secretion by Paneth cells, enhancing mucin production by goblet cells, and promoting IEC migration, thereby limiting inflammation. (For details regarding IL17A see fig 6; for details on IL22 see fig 7.) APCs, antigen presenting cells; DCs, dendritic cells; IECs, intestinal epithelial cells; IL, interleukin; NOD, nucleotide binding and oligomerisation domain; Th, T helper; TLR, toll-like receptor.
TH17 EFFECTOR CYTOKINES AS MEDIATORS OF INTESTINAL INFLAMMATION
Wilson et al demonstrated that Th17 cells are characterised by an increased expression of IL17A, IL17F, IL22, IL26, IFNγ and CCL20.60 Consistently, we and others demonstrated an increased expression of IL17A,78 IL17F,79 IL22,80 81 IL2642 and CCL2082 in active Crohn’s disease, supporting a role for these cytokines and chemokines in the disease pathogenesis. Given that the receptors for IL17, IL22 and IL26 are expressed by intestinal epithelial cells,42 80 83 Th17 cells exert a number of biological functions on the intestinal barrier. An overview of this complex interplay between Th17 cells and the intestinal barrier contributing to the perpetuation of intestinal inflammation in Crohn’s disease is given in fig 5. The specific roles of the main Th17 effector cytokines on the intestinal barrier and their role in intestinal inflammation are discussed in the following sections.
Interleukins 17A and 17F
The main characteristic of Th17 cells is the expression of IL17A and IL17F. Although IL17A is mainly expressed by Th17 cells, other sources of this cytokine including murine CD8+ cells,84 85 natural killer T cells (NKT),86 and γδ T cells87 88 were also identified. IL17A is bound by the IL17 receptor A (IL17RA), which is expressed by epithelial cells, endothelial cells and fibroblasts.89 IL17A activates nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) pathways.83 90 91 IL17-deficient mice have decreased antigen-specific T cell activation and antibody production.92 93 Fujino et al found an increased expression of IL17A mRNA and intracellular protein in the intestinal mucosa of patients with IBD.78 Similarly, we demonstrated increased IL17F mRNA expression levels in patients with active Crohn’s disease.79 In colonic epithelial cells, IL17A differentially regulates chemokine expression (fig 6).94 In a model of trinitrobenzene sulfonic acid (TNBS)-induced colitis, mice deficient in the expression of the IL17 receptor IL17RA showed reduced CXCL2 expression and reduced neutrophil migration into the colon.95 Another recent study demonstrated that IL17A and IL17F have a redundant but highly pathogenic role in a murine model of intestinal inflammation.96 Interestingly, IL17RA-deficient mice also show defective granulocyte colony stimulating factor (G-CSF) responses, and defects in the granulopoiesis resulting in enhanced susceptibility to experimental Klebsiella pneumoniae pulmonary infection,97 suggesting that IL17RA inhibition may increase the risk for infectious complications. While the majority of IL17A-mediated effects increase the inflammatory response (fig 6), some studies indicate epithelial barrier protective responses such as upregulation of antimicrobial peptides98 and maintaining the epithelial barrier by modulating tight junctions.99 Figure 6 gives an overview of the multiple functions of IL17A which have been implicated in the perpetuation of inflammation in Crohn’s disease. Efficacy and safety studies with AIN457, a monoclonal anti-IL17 antibody, are under way for Crohn’s disease.100 The outcomes of this study will help to uncover the potential of specific targeting of IL17 for treating Crohn’s disease.

The multiple effects of the main Th17 cytokine IL17A on the perpetuation of inflammation in Crohn’s disease. IL17A is the main cytokine released by Th17 cells and exerts a number of proinflammatory effects on intestinal epithelial cells, endothelial cells, macrophages, fibroblasts and colonic myofibroblasts through increased release of proinflammatory chemokines, cytokines and other proinflammatory mediators such as prostaglandins, MMPs, NOS-2 and G-CSF. This results in recruitment and accumulation of granulocytes, lymphocytes and DCs and further perpetuation of the inflammatory response by increasing the production of proinflammatory cytokines and chemokines. However, there is also evidence that IL17A (particularly in combination with IL22) may enhance the production of antimicrobial peptides such as defensins in Paneth cells. DCs, dendritic cells; G-CSF, granulocyte colony stimulating factor; GM-CSF, granulocyte macophage colony stimulating factor; IL, interleukin; MMPs, matrix metalloproteinases; MUC, mucin; NOS-2, nitric oxide synthase-2; TNF, tumour necrosis factor.
Interleukin 22
The IL10-like cytokine IL22 was recently shown to be expressed by Th17 cells98 101 102 and in subsets of natural killer (NK) cells.103–107 The IL22 receptor complex is composed of the specific subunit IL22R1 and the IL10R2 subunit, which is also shared with the receptors for IL10, IL26 and the lambda-IFNs (IL28A, IL28B and IL29).42 80 108–111 We recently demonstrated expression of the IL22 receptor complex in intestinal epithelial cells.80 IL22 receptor activation results in activation of STAT3 and increased transcription of proinflammatory cytokines such as IL8 and TNFα80 (fig 7). Similarly, the upregulation of proinflammatory cytokines, chemokines and metalloproteinases mediated via activation of NF-κB and AP-1 has been shown in IL22-stimulated colonic subepithelial myofibroblasts112 (fig 7).

The proinflammatory and epithelial barrier protective properties of the Th17 cytokine IL22 with divergent effects in the pathogenesis of Crohn’s disease. IL22, produced in Th17 cells and subsets of natural killer cells (NKp46), has both proinflammatory properties (mediated via increased secretion of proinflammatory cytokines and chemokines from intestinal epithelial cells and colonic subepithelial myofibroblasts) but also epithelial barrier protective functions mediated by enhancing migration and proliferation of intestinal epithelial cells and the release of antimicrobial peptides from Paneth cells and mucins from goblet cells. AP, activator protein-1; IECs, intestinal epithelial cells; IL, interleukin; LBP, lipopolysaccharide (LPS)-binding protein; LIF, leukaemia inhibitory factor; MMPs, matrix metalloproteinases; MUC, mucin; NF-κB, nuclear factor kappa B; Reg, regenerating islet derived (protein); STAT, signal transducer and activator of transcription; Th, T helper; TNF, tumour necrosis factor.
In addition to its proinflammatory properties, we demonstrated barrier protective functions for IL22 including increased migration and proliferation of intestinal epithelial cells80 which we found in other cell types including hepatocytes.113 114 These data are further supported by the genome-wide search of potential downstream targets in IL22-treated keratinocytes uncovering a large group of genes involved in tissue repair and wound healing.115 116 Moreover, we and others found an increased expression of protective defensins upon stimulation with IL22.80 117 We recently demonstrated that particularly β-defensin-2, which is transcriptionally upregulated by IL22,80 promotes intestinal wound healing.118 In addition to defensins, IL22 increases the expression of a number of other epithelial protective, antimicrobial peptides119 (fig 7).120 IL22 may also exert protective effects in Crohn’s disease by increasing the expression levels of lipopolysaccharide (LPS)-binding protein (LBP), thereby antagonising circulating LPS in these patients.121 A functional role for IL22 in Crohn’s disease is further sustained by the increased IL22 expression levels found in the serum and intestine of patients with active Crohn’s disease.80 81 112 121 However, there is evidence that IL22 may play a role in ulcerative colitis. Andoh et al demonstrated increased IL22 expression in active ulcerative colitis although the expression levels were lower than in Crohn’s disease.112 Similar observations were made by our group.80 Moreover, IL22 exhibited protective effects in a colitis model122 and a mouse model of ulcerative colitis.119 Taken together, IL22 has bi-directional functions in Crohn’s disease.123 While it upregulates a number of proinflammatory mediators, it also mediates intestinal barrier function by increasing the expression of antimicrobial peptides and upregulating proliferation and migration of intestinal epithelial cells (fig 7).
Interleukin 26
In addition to IL17A, IL17F and IL22, human Th17 cells produce IL26,60 62 another IL10-like cytokine, which is not expressed in mice. We demonstrated the expression of the IL26 receptor complex composed of IL20R1 and IL10R2 in intestinal epithelial cells.42 IL26 activates MAP kinases, serine/threonine protein kinase (Akt) and STAT1/342 and the gene transcription of other proinflammatory cytokines. However, in contrast to the proliferative effects of IL22,80 it decreases cell proliferation,42 thereby potentially impairing intestinal wound healing. In inflamed colonic lesions of patients with Crohn’s disease, we found an elevated IL26 mRNA expression that correlated highly with IL8 and IL22 expression levels.42 Immunohistochemical analysis demonstrated IL26 protein expression in colonic T cells including RORγt-expressing Th17 cells with an increased number of colonic IL26-expressing cells in active Crohn’s disease.42 IL26-expressing Th17 cells in the intestinal mucosa of patients with Crohn’s disease were also found in the study by Pene et al.43 Currently, the exact functional role of IL26 in intestinal inflammation remains unclear and studies in murine models are limited by the lack of endogenous IL26 expression in mice.
Interleukin 21
IL21 is another cytokine secreted by Th17 cells.124–126 IL21 expression is induced by IL6 in a STAT3-dependent fashion.125 126 IL21 regulates the differentiation of CD4+ T cells to Th17 cells in an autocrine manner, thereby amplifying the Th17 cell response and inducing its own expression in an autocrine loop (fig 4).124–126 Enhanced IL21 expression was found in Crohn’s disease,127 particularly in lamina propria lymphocytes.128 Interestingly, the Th1 cytokine IL12 enhanced IL21 in normal lamina propria lymphocytes through an IFNγ-independent mechanism, and blocking of IL12 in lamina propria mononuclear cells derived from patients with Crohn’s disease decreased anti-CD3-stimulated IL21 expression.127 Neutralisation of IL21 in cultures of Crohn’s disease lamina propria mononuclear cells decreased phosphorylated STAT4 and T-bet expression, thereby inhibiting IFNγ production.127 IL21 may therefore contribute not only to Th17-mediated inflammation but also to the ongoing Th1 mucosal response in Crohn’s disease.127 Part of the proinflammatory activities of IL21 seem to be mediated by an increased production of matrix metalloproteinases (MMPs) in fibroblasts.129 Interestingly, we recently demonstrated that certain single nucleotide polymorphisms (SNPs) in the IL2/IL21 gene region are in epistasis with IL23R gene variants regarding susceptibility to ulcerative colitis130 (see details below).
The chemokine CCL20
The chemokine CCL20 has been shown to be another effector protein of Th17 cells.60 CCL20 is the ligand to CCR6, which is expressed on the surface of Th17 cells. Therefore, Th17 cell-expressed CCL20 may contribute in an autocrine loop to accumulation of Th17 cells and perpetuation of the intestinal inflammation in Crohn’s disease (figs 5 and 6). Consistently, we recently demonstrated increased CCL20 expression in patients with Crohn’s disease which correlated with IL8 mRNA expression.82 In mice, a neutralising monoclonal antibody directed against CCL20 protected against TNBS-induced colitis.131
THE INFLUENCE OF SUSCEPTIBILITY GENES OF CROHN’S DISEASE ON IL23R SIGNALLING AND TH17 CELL DEVELOPMENT
The role of Th17 cells in the pathogenesis of Crohn’s disease is strongly supported by the results of recent genome-wide association studies implicating IL23R and signal transducers downstream of IL23R in the genetic susceptibility to Crohn’s disease. An overview of the most important susceptibility genes for Crohn’s disease and how they contribute to Th17 cell development is given in the following sections.
Interleukins 23R and 12B
The first genome-wide association study in the North American population demonstrated that an uncommon coding variant in the IL23R gene (rs11209026, c.1142G>A, p.Arg381Gln) confers strong protection against Crohn’s disease, and additional non-coding IL23R variants are independently associated.9 This finding has been confirmed by a number of investigators including our group.132 Importantly, IL23R is also associated to a lesser degree with ulcerative colitis.9 132 In contrast to the majority of IBD susceptibility genes identified so far, the minor allele of the IBD-associated IL23R SNPs may modulate the IBD risk in different directions. While some variants (eg, rs11209026, p.Arg381Gln) decrease IBD susceptibility, others (eg, rs1004819) increase IBD susceptibility. Currently, it is unknown how these IL23R SNPs exactly modulate IBD susceptibility, although the leading hypothesis is that the IL23R signalling pathway contributes to IBD by promoting a pro-inflammatory state133 (fig 8). Our group demonstrated that patients with Crohn’s disease and with IBD risk-increasing IL23R variants have higher IL22 serum levels than patients with IBD risk-decreasing IL23R variants.81 Although IL22 has, in addition to its proinflammatory functions, barrier-protective properties80 119 (fig 7), IL22 serum levels correlate with Crohn’s disease activity,81 121 suggesting that IL22 contributes (likely via IL23R) to the pathogenesis of Crohn’s disease. However, so far it is unknown if IL23R variants also modulate the secretion of other Th17 cytokines. In another recent study, certain IL23R variants (including p.Arg381Gln) were retrovirally transduced into human T cell blasts and functionally characterised by measuring the IL23-induced signal transduction pathways, and IFNγ and IL10 production.134 However, no differences were detected between the genetic variants and wild-type regarding the function of the IL23R chain.134 Patients with Crohn’s disease are more likely to develop other autoimmune diseases including ankylosing spondylitis, psoriasis and rheumatoid arthritis. Interestingly, for these diseases, an association with IL23R has also been established by genome-wide association scans.135–137

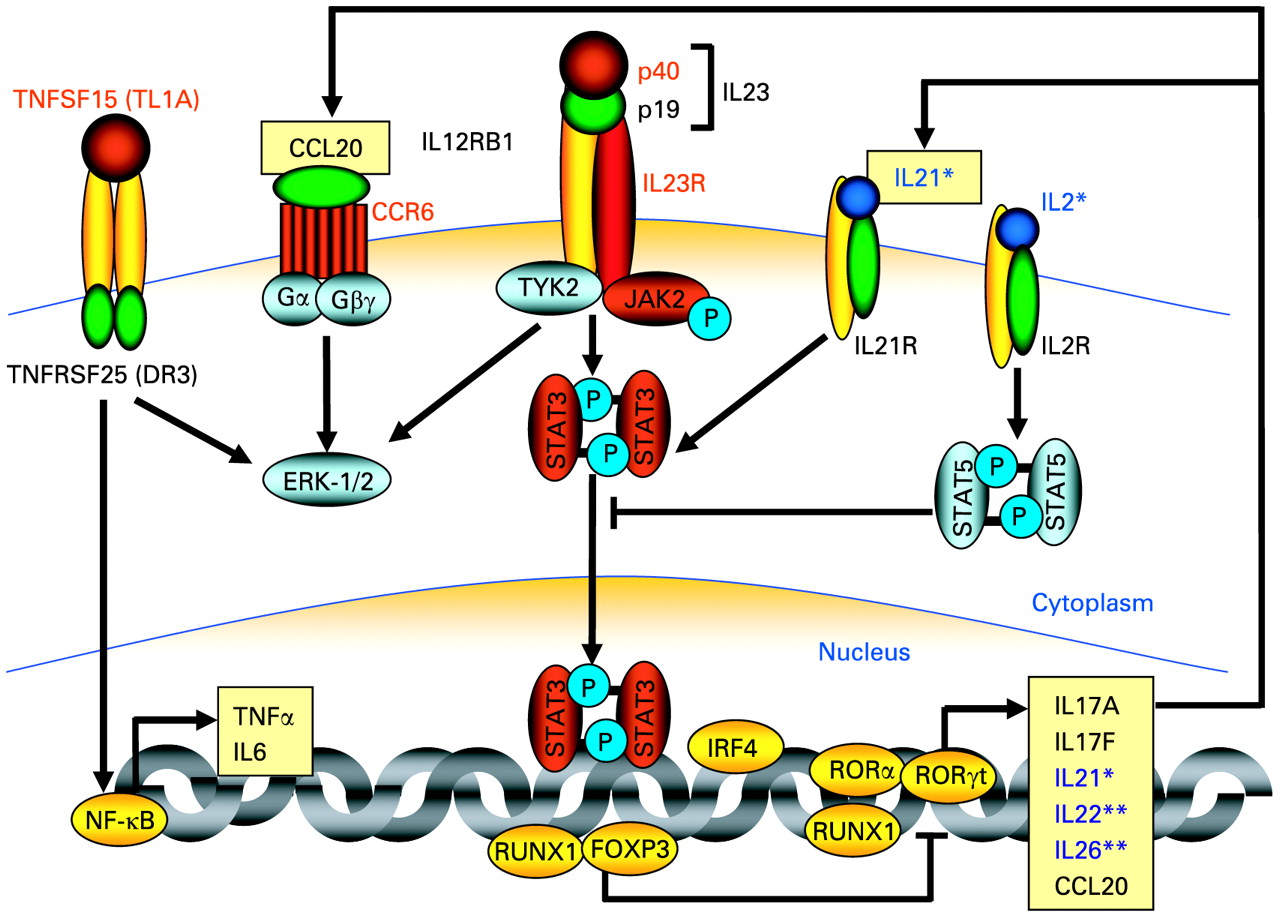
Proteins, which are encoded by susceptibility genes for Crohn’s disease and ulcerative colitis, involved in IL23R signalling and Th17 cell development. The figure highlights genes encoding signalling components in Th17 cells, which were found to be associated with Crohn’s disease and partially also with ulcerative colitis. The proteins encoded by genes found to be associated with Crohn’s disease in genome-wide association studies are highlighted in red. Similarly, the genes encoding IL23R, IL12/23 p40, JAK2, and STAT3 were also found to be associated with ulcerative colitis in genome-wide studies. *Recently, we demonstrated an association of SNPs within the IL2/IL21 genes (proteins marked in dark blue) with ulcerative colitis which are in epistasis with the IL23R gene.130 **A very recent genome-wide scan demonstrated an association with susceptibility to ulcerative colitis for SNPs in and within the proximity of the IL22/IL26 gene cluster.157 ERK, extracellular signal-regulated kinase; FOXP3, forkhead box P3; IL23R, interleukin 23 receptor; IRF, interferon regulatory factor; JAK2, Janus kinase 2; NF-κB, nuclear factor kappa B; ROR, retinoid-related orphan receptor; RUNX, runt related transcription factor; SNP, single nucleotide polymorphism; STAT, signal transducer and activator of transcription; Th, T helper; TNF, tumour necrosis factor; TNFRSF, TNF receptor superfamily 25; TNFSF, TNF superfamily; TYK, tyrosine kinase;
The important role of IL23R in the pathogenesis of Crohn’s disease is further supported by the findings of a large meta-analysis of the first three genome-wide association studies.8 Among the more than 30 regions with the highest association signal with Crohn’s disease, four SNPs are within genes with a major role in IL23R signalling (IL23R, IL12B, STAT3 and JAK2) (fig 8).8 Two other susceptibility genes for Crohn’s disease (CCR6, TNFSF15)8 are also involved in Th17 differentiation (figs 8 and 9). Interestingly, significant disease associations with IL23R, IL12B, STAT3 and JAK2 have also been demonstrated in ulcerative colitis.9–11 132 138 IL12B encodes the IL12/IL23 subunit p40, which is part of both IL12 and IL23 (fig 8) further demonstrating the difficulty in differentiating the association of this susceptibility gene with Th1 versus Th17 signalling components.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The main groups of susceptibility genes for Crohn’s disease and their effect on Th17 cell development. This figure gives an overview of how the main susceptibility genes for Crohn’s disease participate in the development of Th17 cells. The recognition of bacterial products by APCs depends on pattern recognition receptors such as NOD2 and TLR4. Bacterial products are phagocytosed by APCs, a process that, in the terminal ileum, is also highly dependent on the expression of the chemokine receptor CX3CR1,161 which responds to epithelial cell-expressed fractalkine.162 163 Following phagocytosis, bacteria undergo autophagy which is dependent on the autophagy genes ATG16L1 and IRGM. ATG16L1 may also influence interleukin (IL)1β and IL18 secretion in APCs. Activated APCs secrete IL23, IL6 and IL1β, which inhibit Treg development but are necessary for Th17 cell differentiation, while IL12, also secreted by antigen-stimulated APCs, triggers the development of Th1 cells. Th17 cells produce cytokines such as IL17A and IL22, which act on the intestinal barrier stimulating the release of defensins (from Paneth cells), mucins (from goblet cells), and prostaglandins. IL22 also modulates intestinal barrier function. Defensin secretion from Paneth cells is additionally modulated by Crohn’s disease-associated NOD2 and ATG16L1 mutations. On the other hand, β-defensin-2 has also be shown to be a ligand to TLR4164 and CCR6.165 Prostaglandin E2 binds to its receptor PTGER4 and, together with bacterial products (recognised by pattern recognition receptors such as NOD2 and TLR4), promotes the release of IL23 from dendritic cells. PTGER4 expression-modulating single nucleotide polymorphisms (SNPs) are also associated with Crohn’s disease. PTGER4 is essential for maturation and migration of certain APC subpopulations. SNPs in or within proximity of MUC19 encoding mucins and ICOSL, the ligand for ICOS, have also been shown to be associated with susceptibility to Crohn’s disease. Furthermore, the Crohn’s disease susceptibility gene TNFSF15 encodes TL1A, which is a tumour necrosis factor-like molecule expressed by intraepithelial lymphocytes which binds to DR3 (TNFRSF25) and mediates strong co-stimulation of Th1 and Th17 cells. Genes marked with one asterisk were confirmed in the meta-analysis by Barrett et al.8 The gene marked with two asterisks (TLR4) has been shown to be a Crohn’s disease susceptibility gene in a large meta-analysis.166 APCs, antigen presenting cells; DR3, death receptor 3; ICOS, inducible co-stimulatory molecule; IL, interleukin; NOD, nucleotide binding and oligomerisation domain; PTGER4, prostaglandin receptor EP4; SNP, single nucleotide polymorphism; Th, T helper; TL1A, TNF-like molecule 1A (TNF, tumour necrosis factor); TLR, toll-like receptor; TNFRSF25, TNF receptor superfamily 25; Treg, regulatory T cell.
STAT3 and JAK2
STAT3 is not only a downstream signalling component of IL23 but is also activated by a number of other cytokines including IL6, IL10, IL21, IL22, IL26 and IL31,42 80 113 114 139 suggesting a more complex role of STAT3 in the pathogenesis of IBD which is further supported by both pro- and anti-inflammatory properties of STAT3. This bi-directional effect of STAT3 in intestinal inflammation is supported by the fact that activated STAT3 has been found in human IBD and murine colitis models,140 141 while STAT3 deletion during haematopoiesis causes a Crohn’s-like disease in mice.142 An earlier study, which was performed before establishing the concept of Th17 cells, demonstrated that STAT3 deficiency in macrophages and neutrophils results in enhanced Th1 activity and spontaneous enterocolitis in mice.143 However, a recent study showed that mutations in STAT3 impair the development of human Th17 cells.144 Similar to STAT3, JAK2, a gene encoding an IL23R signalling component up-stream of STAT3, has been shown to be associated with Crohn’s disease8 but also with ulcerative colitis.10
The chemokine receptor CCR6
CCR6, the gene encoding the chemokine receptor CCR6, has been identified in the meta-analysis by Barrett et al as another susceptibility gene of Crohn’s disease.8 CCR6 is the receptor for the chemokine CCL20, for which we demonstrated an increased expression in active Crohn’s disease.82 A marked increase in the number of CCR6-bearing lamina propria CD4+ and CD8+ T cells was observed in TNBS-treated animals,131 implicating CCR6+ T cells as mediators of intestinal inflammation. Several studies indicated CCR6 as a surface marker of Th17 cells,43 45 145–149 suggesting that the genetic susceptibility of Crohn’s disease modulated by variants in the CCR6 gene may be mediated via modulation of Th17 function (fig 8). However, the functional consequences of CCR6 gene variants associated with Crohn’s disease have not been studied yet.
TNFSF15
TNFSF15 encodes TL1A, which is a TNF-like molecule that mediates co-stimulation of Th1 and Th17 cells. Several studies, including the meta-analysis of genome-wide association studies, confirmed TNFSF15 as susceptibility gene for Crohn’s disease.8 150 151 Expression of TL1A is increased in the mucosa of Crohn’s disease patients and murine models of ileitis.152–154 An anti-TL1A antibody prevented chronic colitis and attenuated established colitis by downregulation of both Th1 and Th17 activation155 (figs 8 and 9). TL1A-deficient DCs exhibit reduced capacity in supporting Th17 differentiation and proliferation.156 Therefore, TL1A is required for the optimal differentiation as well as effector function of Th17 cells (fig 8).156
Interleukins 17A, 17F, 22 and 26
The meta-analysis of the first genome-wide association studies did not reveal any SNPs within the IL17A and IL17F genes among the most significantly associated Crohn’s disease susceptibility loci. However, there are some studies demonstrating certain effects of these genes on the susceptibility and phenotype of IBD. A Japanese study demonstrated an influence of certain IL17A and IL17F SNPs on ulcerative colitis susceptibility and the ulcerative colitis phenotype.158 However, our group did not observe significant differences regarding genotype frequencies of the IL17F p.His161Arg SNP between healthy controls and IBD patients.79 A North American study demonstrated an interaction between IL23R and IL17A and between IL23R and IL17RA haplotypes regarding susceptibility to Crohn’s disease.159 They found associations of IL17RA with both Crohn’s disease and ulcerative colitis, while IL17A appeared to be specific for Crohn’s disease.160 A very recent genome-wide association study demonstrated an association of a SNP within the IL26 gene with susceptibility to ulcerative colitis, while other SNPs within proximity to the IL22/IL26 gene cluster were also associated with ulcerative colitis;171 however, their effect in Crohn’s disease is currently unclear.
Interleukins 2 and 21
IL2 and IL21 may also influence IL23 signalling (fig 8). While IL21 synergises with IL23 to activate STAT3 and Th17 cell differentiation, IL2 inhibits STAT3 signalling (and Th17 cell development) via STAT5 activation, thereby reciprocally favouring Treg development (figs 4 and 8). We recently demonstrated that SNPs within the KIAA1109/TENR/IL2/IL21 linkage disequilibrium block on chromosome 4q27 had a protective effect on susceptibility to ulcerative colitis.130 Importantly, in ulcerative colitis, epistasis was observed between the IL23R SNP rs1004819 and three SNPs in the KIAA1109/TENR/IL2/IL21 block (rs13151961, rs13119723 and rs6822844).130 Interestingly, SNPs within the chromosome 4q27 region were also found to be associated with other autoimmune diseases including coeliac disease, rheumatoid arthritis, type 1 diabetes, Graves’ disease and psoriatic arthritis,136 167–169 suggesting a common genetic background for these autoimmune diseases.
NOD2/CARD15, TLR4 and TLR9
NOD2/CARD15 is the most important and most often replicated susceptibility gene for Crohn’s disease which was also confirmed in the first meta-analysis of genome-wide association studies.8 Interestingly, DCs activated by the NOD2 ligand muramyl dipeptide (MDP) induce the production of IL17A and promote Th17 cell development via IL23 secretion (fig 9).170 DCs with Crohn’s disease-associated NOD2 mutations have reduced capacity to induce enhanced levels of IL17A when primed with TLR ligands.170 Similarly, we recently demonstrated that monocytes from individuals with two mutated NOD2 alleles displayed an impaired release of TNFα and IL10 but also of IL1β and IL12/IL23p40 in response to MDP.171 Interestingly, monocytes from patients with Crohn’s disease with two mutated NOD2 alleles displayed significantly higher basal levels of IL12/IL23p40 in cell culture supernatants compared to wild-type Crohn’s disease patients and control individuals,171 suggesting increased baseline IL12/IL23 activity in patients with NOD2 mutations. The increased baseline IL12/IL23 activity could be an underlying mechanism of the chronic mucosal inflammation seen in Crohn’s disease, particularly in patients with two NOD2 mutations, especially the p.Leu1007fsX1008 variant.172 173 Therefore, NOD2 mutations may contribute to DC dysfunction,170 resulting in Crohn’s disease although a number of other mechanisms including decreased defensin production are under consideration (fig 9).174 175 Consistent with the reduced defensin expression, intestinal epithelial cells expressing mutated NOD2 have a reduced capacity to restrict proliferation of bacterial pathogens in monolayer cultures,176 raising the possibility that NOD2 mutations may increase the bacterial colonisation of the intestine resulting in increased DC activation and IL23-driven Th17 responses.
In a large meta-analysis, gene variants in TLR4, another pattern recognition receptor which mediates LPS signalling, were found to be associated with Crohn’s disease but not with ulcerative colitis.166 However, this finding has not yet been confirmed in genome-wide association scans. In addition, we demonstrated that the TLR4 p.Asp299Gly and p.Thr399Ile variants were particular strong predictors for a stricturing phenotype in Crohn’s disease,177 TLR4 activation induces both IL12/IL23p40 and IL12p35 mRNA expression,178 suggesting that bacterial LPS may trigger both Th1 and Th17 immune responses.
We recently demonstrated epistasis between TLR9 SNPs and IL23R SNPs as well as NOD2 mutations regarding susceptibility to Crohn’s disease,179 suggesting that TLR9, a gene encoding a receptor for bacterial DNA, modulates IL23R-mediated effects on susceptibility to Crohn’s disease. Of interest, Tlr9−/− mice display increased frequencies of Treg cells within intestinal effector sites and reduced constitutive Th17 and Th1 effector cells (Teff).182 These data complement the recent findings by Dan Littman’s group74 and suggest that commensal DNA is a natural adjuvant for priming intestinal responses via modulation of the Treg/Teff cell equilibrium.180
Key points
Th1 and Th17 cells in Crohn’s disease
Both T helper (Th)1 and Th17 cytokines are upregulated in Crohn’s disease, while the function of tolerising, anti-inflammatory regulatory T cells (Tregs) is impaired.
In addition to Th1 and Th17 cells, a T cell population sharing features of both Th1 and Th17 cells (“Th17/Th1 cells”) may play an additional role in Crohn’s disease.
However, many activities previously ascribed to the Th1 cytokine interleukin (IL)12 may have been mediated by IL23, an activator of Th17 cells, given that IL12/IL23p40 is part of both cytokines.
Differentiation of Th17 cells
IL1β, IL6, transforming growth factor (TGF)β, IL21 and IL23 are key cytokines involved in the differentiation of Th17 cells.
TGFβ reciprocally regulates the differentiation of inflammatory Th17 cells and suppressive Treg subsets. The concomitant presence of proinflammatory cytokines favours Th17 cell differentiation, while IL2 and retinoic acid promote Treg development.
Bacteria and probiotics modulate Th17 cell development.
Effector cytokines of Th17 cells
IL17A, IL17F, IL21, IL22, IL26 and CCL20 are the main effector cytokines and chemokines of human Th17 cells. Their transcription is mediated via retinoid-related orphan receptor (ROR)γt, the key transcription factor in Th17 cells.
Receptors for most Th17 cytokines are expressed on intestinal epithelial cells. Th17 cytokines perpetuate the intestinal inflammation by further increasing the expression of proinflammatory cytokines and chemokines.
Genetic susceptibility to inflammatory bowel disease and IL23 signalling
Genes encoding proteins involved in downstream signalling of IL23 and Th17 cell differentiation (IL23R, IL12B, JAK2, STAT3, CCR6 and TNFSF15) are associated with susceptibility to Crohn’s disease and also partly to ulcerative colitis. Single nucleotide polymorphisms (SNPs) in and within the proximity of the IL22/IL26 gene cluster are associated with ulcerative colitis.
Other susceptibility genes (eg, NOD2, ATG16L1 and PTGER4 expression-modulating SNPs in 5p13.1) may also directly or indirectly modulate Th17 responses.
Anti-IL12/IL23 and anti-IL17 treatment strategies in Crohn’s disease
An anti-IL12/IL23p40 monoclonal antibody that neutralises IL12 and IL23 demonstrated clinical efficacy in patients with Crohn’s disease.
An anti-IL23p19 monoclonal antibody, which targets only IL23, was effective in a murine colitis model and clinical studies in Crohn’s disease are anticipated.
A phase 2 trial investigating a monoclonal antibody against IL17A in patients with moderate to severe active Crohn’s disease is ongoing.
ATG16L1, IRGM, and PTGER4 expression modulating SNPs in 5p13.1
With the exception of NOD2 and IL23R, ATG16L1 is the most commonly replicated susceptibility gene for Crohn’s disease.8 181–183 Similar to IRGM, another susceptibility gene for Crohn’s disease,184 ATG16L1 is essential for autophagy of bacteria.182 185 In addition, knockout of ATG16L1 results in Paneth cell dysfunction186 and enhances endotoxin-induced IL1β production187 (fig 9). So far, a direct influence of autophagy genes on Th17 cell development has not been shown, although a defect autophagy of bacteria may influence APC function, which could modulate production of essential cytokines required for Th17 differentiation such as demonstrated for IL1β.187
Another group of susceptibility loci found to be associated with Crohn’s disease relates to prostaglandin E2 signalling. A Belgian–French genome-wide association study demonstrated that certain SNPs in a gene desert on chromosome 5p13.1 are strongly associated with susceptibility to Crohn’s disease;188 specifically, this study found an association between SNPs in this chromosomal region modulating the expression of the prostaglandin receptor EP4 (PTGER4) with the disease susceptibility.188 We recently confirmed that SNPs in the 5p13.1 region associated with PTGER4 expression are associated with Crohn’s disease.189 Importantly, we found evidence for strong epistasis between this region and several SNPs within the ATG16L1 gene regarding susceptibility to Crohn’s disease.189 Currently, it is unknown how these SNPs in the 5p13.1 region influence the pathogenesis of Crohn’s disease. However, it is likely that these effects are mediated via the PTGER4 ligand prostaglandin E2 and via DC dysfunction190 (fig 9). It may be hypothesised that such a DC dysfunction could be further exacerbated by defects in the autophagy pathway; eg, by mutations in ATG16L1, which would explain the strong epistasis between the 5p13.1 region and ATG16L1 found in our study.189 Importantly, a recent study showed that the proinflammatory effect of prostaglandin E2 in an experimental IBD model is mediated through the IL23–IL17 axis.191 In the presence of exogenous prostaglandin E2, peripheral blood mononuclear cells produced higher IL17A, CCL20 and CXCL8, and lower IFNγ levels than in control cultures.192 Exogenous prostaglandin E2 and IL23 synergised in inducing IL17A.192 However, prostaglandin E2 has also been implicated in the induction of Tregs193 and in their suppressive capacity194 suggesting that, similar to TGFβ, prostaglandin E2 may have a bi-directional influence on T cell differentiation depending on the predominant cytokine environment. Figure 9 summarises how the different susceptibility genes for Crohn’s disease may collectively contribute to the pathogenesis of Crohn’s disease.
ULCERATIVE COLITIS: ANOTHER TH17-MEDIATED DISEASE?
There is good evidence that ulcerative colitis is characterised by an exaggerated Th2-like response as demonstrated by increased production of Th2 cytokines such as IL5 and IL13 in this disease.3 5 19 However, the fact that five genes involved in downstream signalling of IL23 (IL23R, IL12B, JAK2, STAT3, IL26) mediate susceptibility to ulcerative colitis suggests strongly that Th17 cells are also involved in its pathogenesis.9–11 132 157 Previously, it has been assumed that Th17 cells do not contribute to the pathogenesis of Th2 disorders since IL4 and IL25 are both potent inhibitors of Th17 development.195 However, Kobayashi et al demonstrated a significant upregulation of IL17A in lamina propria CD4+ T cells following IL23 stimulation in ulcerative colitis.41 Upregulated IL23p19 mRNA expression correlated with IL17A.41 Similarly, we and others found high expression levels of the Th17 cytokines IL17A, IL22 and IL26 not only in inflamed colonic lesions of patients with Crohn’s disease but also in active ulcerative colitis.42 78 80 112 Taken together, these findings suggest that Th17 cells also modulate the predominant Th2-mediated inflammation in ulcerative colitis.
CONCLUSIONS
The previous Th1/Th2 paradigm indicating predominant Th1-mediated responses in Crohn’s disease and an exaggerated Th2-like inflammation in ulcerative colitis has to be revised and updated with the discovery of Th17 cells and Treg cells. While Th17 cells contribute to intestinal inflammation in both forms of IBD, particularly in Crohn’s disease, Treg cells have tolerising, anti-inflammatory properties. The results of the recent genome-wide association studies, implicating genes encoding downstream signalling components of IL23 signalling in the disease susceptibility of Crohn’s disease and ulcerative colitis, clearly indicate that IL23R-expressing Th17 cells may play a role in both forms of IBD.
Given that both the Th1 cytokine IL12 and the Th17 modulator IL23 contain the p40 cytokine subunit, a number of inflammatory activities previously ascribed to IL12 may have been mediated by IL23 and the role of Th1 cells in the pathogenesis of Crohn’s disease may have been overestimated. Nevertheless, the current evidence suggests that both Th1 and Th17 cells contribute to intestinal inflammation in Crohn’s disease, in which even a third subpopulation of T cells (“Th17/Th1 cells”), sharing features of both Th1 and Th17 cells, can be found. Key cytokines involved in the differentiation of Th17 cells are IL1β, IL6, TGFβ, IL21 and IL23. TGFβ reciprocally regulates Treg differentiation under the influence of IL2 and retinoid acid, while it favours Th17 cell development in the concomitant presence of proinflammatory cytokines. RORγt is the key transcription factor for Th17 cells. Bacteria and probiotics may modulate Th17 cell development. Considering that IL23 and IL12, two cytokines essential for Th17 and Th1 development, are produced in APCs after contact with bacteria and bacterial products, it is likely that the exaggerated T cell response in Crohn’s disease is rather a secondary result caused by a primary barrier defect in genetically susceptible individuals with subsequent bacterial invasion and APC activation.
IL17A, IL17F, IL21, IL22, IL26 and CCL20 are the main effector cytokines and chemokines of human Th17 cells. For most of these cytokines, receptors are expressed on intestinal epithelial cells and exposure to Th17 cytokines exaggerates the inflammatory response by amplifying the secretion of proinflammatory cytokines, chemokines, MMPs and prostaglandins. Th17 cells also secrete some amounts of TNFα and IL6. Recent evidence suggests that anti-TNF treatment strategies inhibit inflammatory DC cytokine production and maturation, leading to reduced Th17 cell activity.196 An anti-IL12/23p40 antibody, which inhibits both IL12-triggered Th1 inflammation and IL23-mediated Th17 responses, has been successfully applied in patients with Crohn’s disease.36 37 However, the clear contributions of inhibiting Th1 and Th17 responses in Crohn’s disease can only be analysed with an antibody inhibiting the IL23 specific p19 subunit which demonstrated anti-inflammatory properties in a murine colitis model.30 The effector cytokines IL17A, IL17F, IL21, IL22 and IL26, and the chemokine CCL20 are potential future therapeutic targets. Studies targeting IL17 in Crohn’s disease are on the way,100 and an anti-CCL20 antibody demonstrated clinical efficacy in a murine colitis model.131 However, IL22, in particular, seems to mediate both proinflammatory as well as epithelial barrier protective responses80 and murine models of inflammation suggested even anti-inflammatory, protective effects of IL22 in colitis.119 122 Therefore, further understanding and characterisation of the IL23–IL17 pathway and the numerous biological effects it mediates is needed. Novel treatment strategies have to clarify if it is necessary to inhibit the entire IL23/Th17 axis through blockade of IL23, or if it is more efficacious and safer to target select Th17 cell products such as IL17A.
REFERENCES
Footnotes
Competing interests: None.
Funding: SB was supported by grants of the Deutsche Forschungsgemeinschaft (BR 1912/5–1), Else Kröner-Fresenius-Stiftung (Else Kröner-Fresenius Memorial Grant 2005; 60/05//EKMS 05/62), the Ludwig Demling Grant 2007 of DCCV e.V., and a grant of the Excellence Initiative of Ludwig-Maximilians-University Munich (Investment Fund 2008).