Article Text

Abstract
Background and aim: The present study was conducted to examine the effect of activin A on activation of rat pancreatic stellate cells (PSCs).
Methods: PSCs were prepared from rat pancreas using collagenase digestion and centrifugation with Nycodenz gradient. Activation of PSCs was examined by determining smooth muscle actin expression with western blotting. The presence of activin A receptors in PSCs was investigated by reverse transcription-polymerase chain reaction (RT-PCR), western blotting, and immunocytochemistry. Expression of activin A and transforming growth factor β (TGF-β) mRNA was examined by RT-PCR. Activin A and TGF-β peptide concentrations were examined with ELISA. Existence of activin A peptide in PSCs was investigated by immunocytochemistry. Collagen secretion was determined by Sirius red dye binding.
Results: Activin A receptors I and IIa were present in PSCs. PSCs expressed activin A mRNA and secreted activin A. Activin A enhanced PSC activation and collagen secretion in a dose dependent manner. TGF-β and activin A increased each other’s secretion and mRNA expression of PSCs. Follistatin decreased TGF-β mRNA expression and TGF-β secretion of PSCs, and inhibited both PSC activation and collagen secretion.
Conclusion: Activin A is an autocrine activator of PSCs. Follistatin can inhibit PSC activation and collagen secretion by blocking autocrined activin A and decreasing TGF-β expression and secretion of PSCs.
- pancreatic stellate cell
- activin A
- follistatin
- pancreatic fibrosis
- PSC, pancreatic stellate cell
- HSC, hepatic stellate cell
- α-SMA, α smooth muscle actin
- TGF-β, transforming growth factor β
- RT-PCR, reverse transcription-polymerase chain reaction
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
Statistics from Altmetric.com
- PSC, pancreatic stellate cell
- HSC, hepatic stellate cell
- α-SMA, α smooth muscle actin
- TGF-β, transforming growth factor β
- RT-PCR, reverse transcription-polymerase chain reaction
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
Activin A is a homodimeric protein with a molecular mass of 25 kDa, originally isolated from ovarian fluid as a stimulant for follicle stimulating hormone secretion.1 Based on its molecular structure, activin A is considered to be a member of the transforming growth factor β (TGF-β) superfamily.2 Subsequent studies have revealed diverse actions of activin A on cellular growth and differentiation in various types of cells. Activin A increases the number of gonadotrophs in anterior pituitary cultures3 and modulates differentiation of ovarian granulosa cells.4 Activin A also induces differentiation of erythroid cells5 and megakaryocytes,6 and inhibits hepatocyte proliferation.7 In the pancreas, activin A is present in islets A and D cells8 and regulates B cell differentiation.9 Follistatin is an endogenous activin A binding protein known to block the effect of activin A.10 For instance, follistatin protected the activin inhibitory action on hepatocyte proliferation in vitro,7 and overexpression of follistatin in epidermis of transgenic mice impaired wound healing which is controlled by activin A.11 Thus follistatin can be used as a potent activin A blocker both in vitro and in vivo.
Pancreatic stellate cells (PSCs) have been recently identified, isolated, and characterised.12,13 In the normal pancreas, PSCs possess fat droplets containing vitamin A and are quiescent, defined by desmin positive but α smooth muscle actin (α-SMA) negative staining.12 When cultured in vitro, PSCs are autoactivated (autotransformed) changing their morphological and functional features.13 PSCs commence losing vitamin A containing lipid droplets, are highly proliferative, with increasing expression of α-SMA, and produce and secrete extracellular matrix components such as collagen and fibronectin. Namely, PSCs are autotransformed to myofibroblast-like cells. In vivo, PSCs are also activated during both human and experimental pancreatic fibrosis.14 Therefore, PSCs are believed to play an important role in pancreatic fibrogenesis.
TGF-β is one of the major profibrogenic cytokines in various tissues. Recently, evidence of TGF-β participation in pancreatic fibrogenesis has been found. Transgenic mice overexpressing TGF-β in islet cells develop fibrosis of the exocrine pancreas. Moreover, inhibition of TGF-β by anti-TGF-β antibody reduced extracellular matrix production in rat cerulein pancreatitis.15 TGF-β has also been shown to promote PSC activation and extracellular matrix production in an autocrine manner. TGF-β secreted from cultured PSCs controls PSC growth and promotes their activation and extracellular matrix production.16,17 In human chronic pancreatitis tissue, TGF-β expression was observed in acinar cells adjacent to areas of fibrosis and in spindle cells in fibrotic bands.14 Thus TGF-β is assumed to promote pancreatic fibrosis, in part by activating PSCs and enhancing their extracellular matrix production. Although activin A is also a member of the TGF-β family and its physiological role in pancreatic cellular differentiation has been well described,9,18 its pathophysiological role in pancreatic disorders is still unknown. As activin A modulates cell growth and differentiation of various cells in a variety of organs, it is an interesting possibility that activin A also modifies PSC function and participates in pancreatic fibrosis.
In the present study, we examined the effect of activin A on PSC function. We report here that activin A activates PSCs and collagen secretion. Furthermore, activin A is released from PSCs, and activin A and TGF-β enhance each other’s secretion and mRNA expression of PSCs. Finally, follistatin blocks PSC activation and collagen secretion by blocking activin A and reducing TGF-β release and mRNA expression of PSCs. Hence activin A is an autocrine activator of PSCs and follistatin can be used potentially in the treatment of pancreatic fibrosis.
METHODS
Materials
Recombinant human activin A and rabbit antiactivin A polyclonal antibody were kindly provided by Dr Yuzuru Eto (Ajinomoto Co., Japan). Pronase, Nycodenz, and anti-SMA antibody were purchased from Sigma (St Louis, Missouri, USA). DNase I was from Roche (Basel, Switzerland). Collagenase P was from Boehringer Mannheim (Mannheim, Germany). Follistatin was from R&D Systems (Abington, UK). Antiactivin receptor type I and type IIa antibodies were from Santa Cruz (Santa Cruz, California, USA). Horseradish conjugated donkey antimouse IgG, Cy3 conjugated donkey antirabbit IgG, Cy3 conjugated donkey antigoat IgG, and FITC conjugated donkey antimouse IgG antibodies were from Jackson Immuno Research (West Grove, Pennsylvania, USA).
Isolation and culture of rat pancreatic stellate cells
Rat pancreatic stellate cells were prepared as described previously.12 Briefly, rat pancreas was digested in Gey’s balanced salt solution supplemented with 0.05% collagenase P, 0.02% pronase, and 0.1% DNase I. After filtration through nylon mesh, cells were centrifuged in a 13.2% Nycodenz gradient at 1400 g for 20 minutes. PSCs in the band just above the interface of the Nycodenz solution and the aqueous one were collected, washed, and resuspended in Iscove’s modified Dulbecco’s medium containing 10% fetal calf serum, 100 U/ml penicillin, and 100 mg/ml streptomycin. PSCs were cultured in a 5% CO2 atmosphere at 37°C. All experiments were carried out using PSCs between passages one and two.
Western blotting
Western blotting was carried out as described previously,19 using the enhanced chemiluminescence reagent to visualise the secondary antibody. For gel electrophoresis, 10 µg of protein were loaded on each lane of a 10% sodium dodecyl sulphate-polyacrylamide gel.
Immunofluorescence microscopy
Immunofluorescence microscopy was performed as described previously20 using an Olympus BX51 microscope (Olympus Co., Tokyo, Japan). Images were digitised and then processed using Photoshop 5.0 software (Adobe Systems Inc., Mountain View, California, USA).
Quantification of activin A and TGF-β
Concentrations of activin A and TGF-β in culture media of PSCs were determined using commercial kits from R&D Systems and DRG International (Mountainside, New Jersey, USA), respectively, according to the manufacturer’s instructions.
Quantification of collagen secretion
Collagen secreted into culture medium by PSCs was determined using Sircol Sirius red dye (Biocolor Ltd, Newtownabbey, UK), as described previously.21 Collagen was measured by spectrophotometry at 540 nm.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from PSCs with TRIzol reagent (Life Technologies BRL, Grand Island, New York, USA). First strand cDNA was made from total RNA using ReverTra Ace system (Toyobo, Tokyo, Japan) according to the manufacturer’s instructions. PCR for TGF-β was carried out using the PCR kit for rat TGF-β (Maximbio, San Francisco, California, USA). PCR for rat activin A βA subunit, rat activin type I receptor, rat activin type IIa receptor, and rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was performed with the following primers:
-
rat activin A βA subunit: sense 5′-GGACCTAACTCTCAGCCAGAGATG-3′, antisense 5′-TCTCAAA-ATGCAGTGTCTTCCTGG-3′;
-
rat activin type I receptor: sense 5′-GGTCTAT-GAGCAGGGGAAGATGAC-3′, antisense 5′-ACATTTTCGCCTTGCCAGC-3′;
-
rat activin type IIa receptor: sense 5′-AGATGGAAGTCACACAGCCCAC-3′, antisense 5′-CAACACTGGTGCCTCTTTTCTCTG-3′;
-
rat GAPDH: sense 5′-CATGACCACAGTCCATGCCATC-3′, antisense 5′-CACCCTGTTGCTGTA-GCCATATTC-3′.
The reactions were conducted in a DNA Thermal Cycler (Perkin-Elmer Corp., Norwalk, Connecticut, USA) with the following cycle conditions: denaturation at 94°C for one minute, annealing at 52°C for 45 seconds, and extension at 72°C for 45 seconds. The number of cycles was 30 for rat activin A βA subunit, TGF-β, and rat activin receptors, and 18 for GAPDH.
Statistical analysis
ANOVA was used to determine statistical significance. A p value of <0.05 was considered significant.
RESULTS
Activin receptors I and IIa are present in PSCs
As the first attempt to elucidate activin A effects on PSCs, we examined the presence of activin A receptors in PSCs. As activin receptors function as a heterodimer of type I and type II receptors, we examined the presence of both activin receptor type I and type IIa. As shown in fig 1A, RT-PCR with primers specific for rat activin receptor I and IIa amplified 480 (activin receptor I) and 423 (activin receptor IIa) base pair sequences, respectively. These PCR products were verified by sequencing (data not shown). These data indicate that mRNAs of both activin receptors I and IIa are expressed in PSCs. To confirm expression of activin receptor I and IIa proteins, we carried out western blotting of PSC lysate using antiactivin receptor I and IIa antibodies. As shown in fig 1B, western blotting revealed expression of both activin receptor I and IIa proteins in PSCs. As an independent test of the presence of activin receptors, we applied immunocytochemistry using antiactivin receptor I and IIa antibodies. As shown in fig 2, immunocytochemistry revealed that both signals of activin type I and type IIa receptors are observed in PSCs. These signals were abolished when antibodies were preincubated with competing peptides to each antibody (data not shown). These data indicate that activin receptor components essential in forming functional activin receptor heterodimers are present in PSCs.

Expression of activin receptors I and IIa in pancreatic stellate cells (PSCs). (A) Reverse transcription-polymerase chain reaction of activin receptors I (lane 1) and IIa (lane 2) was carried out using PSC total RNA (1 µg) as a template. The right lane is a size marker of 1 kb DNA ladder. The bands were confirmed by sequencing. (B) Western blotting of activin receptors I (lane 1) and IIa (lane 2) was performed using their specific antibodies. Molecular markers are indicated on the right.

Fluorescence micrographs showing the immunoreactivity of (A) activin receptor type I and (B) activin receptor type IIa in pancreatic stellate cells, with corresponding Nomarski images (C, D). Bar 40 µm.
Activin A activates PSCs and increased collagen secretion
We next examined the effect of activin A on PSC activation and collagen secretion. As shown in fig 3A, activin A increased the amounts of α-SMA in PSCs in a dose dependent manner. The maximum increase was observed at 1 nM. Activin A also enhanced collagen secretion by PSCs (fig 3B). These data indicate that activin A activates PSCs and suggest activin A participation in pancreatic fibrosis.

Effect of activin A on α smooth muscle actin (α-SMA) expression and collagen secretion by pancreatic stellate cells (PSCs). (A)Top panel: western blotting of α-SMA in total homogenates of PSCs was carried out after 48 hours of incubation with the indicated amounts of activin A. Activin A increased α-SMA expression of PSCs. Bottom panel: the blots were reprobed with anti-α-tubulin antibody. Expression of α-tubulin protein, examined as an internal control, was not altered by activin A. (B) Collagen secretion by PSCs into culture medium during 48 hours of incubation with the indicated amounts of activin A was quantified with Sirius red dye. Results are expressed as per cent collagen concentration of controls. Values are means (SEM) of three independent experiments (*p<0.05).
Activin A is secreted from PSCs
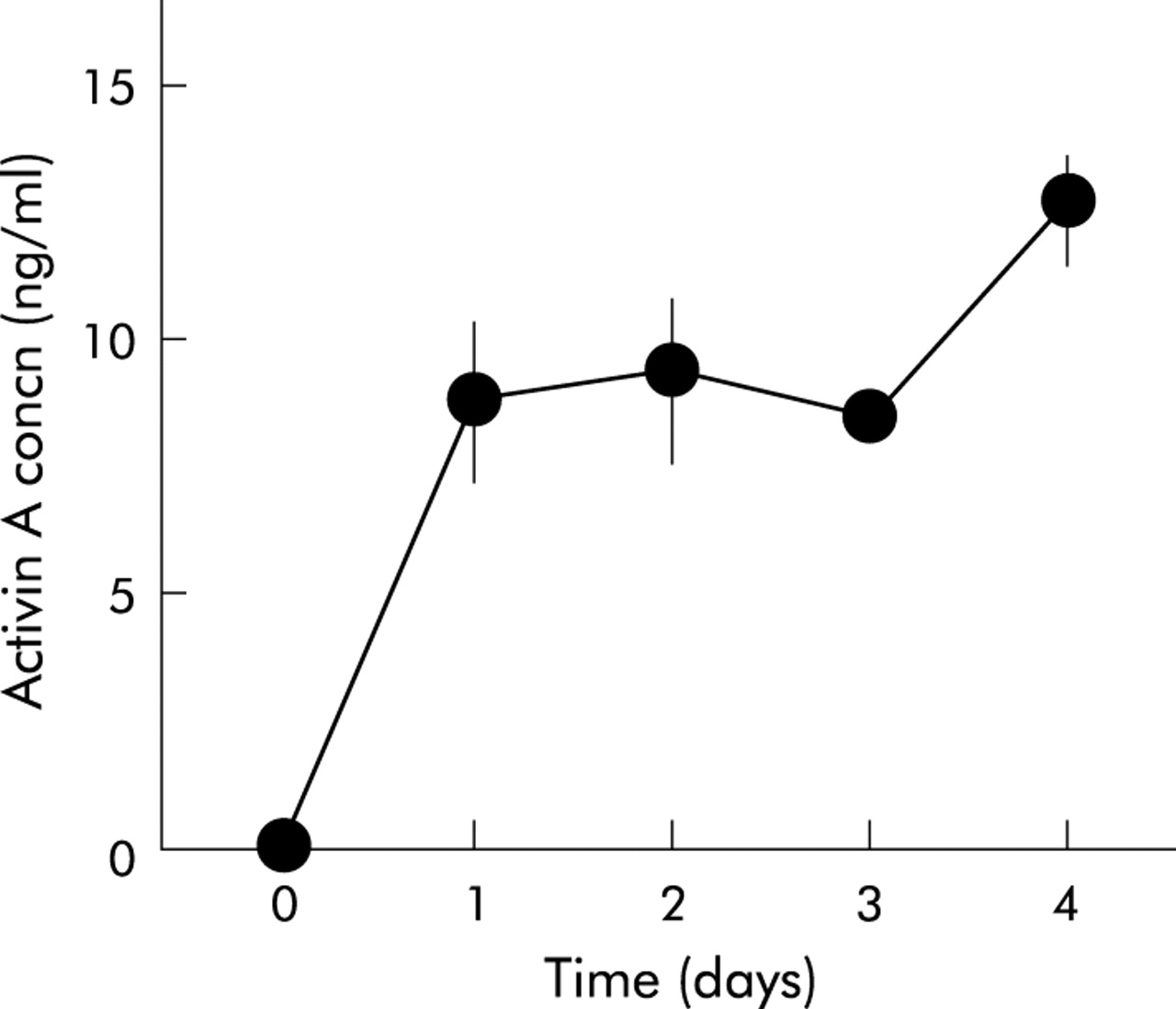
As activin A exerts its effect in an autocrine manner in some cell types,7,22 we examined whether activin A is secreted from PSCs. As shown in fig 4, activin A peptide was detected in PSC culture medium. In particular, activin A concentration in PSC culture medium markedly increased on the first day after the culture medium was changed to fresh medium. No activin A activity was detected in fresh culture medium. These data indicate that activin A is secreted from PSCs. As an independent test of secretion of activin A from PSCs, we examined the existence of activin A peptide in PSCs with immunocytochemistry. We applied double staining with an antiactivin A polyclonal antibody and an anti-α-SMA monoclonal antibody, a marker of activated PSCs. As depicted in fig 5, activin A signals were observed as small dots in PSCs in which α-SMA signals were detected as fine linear architecture. These data indicate that activin A peptide is present in PSCs, confirming that activin A is secreted from PSCs.

Activin A secretion from pancreatic stellate cells. Activin A concentration in culture medium was determined 1–4 days after the culture medium was changed. Results are means (SEM) of three independent experiments.
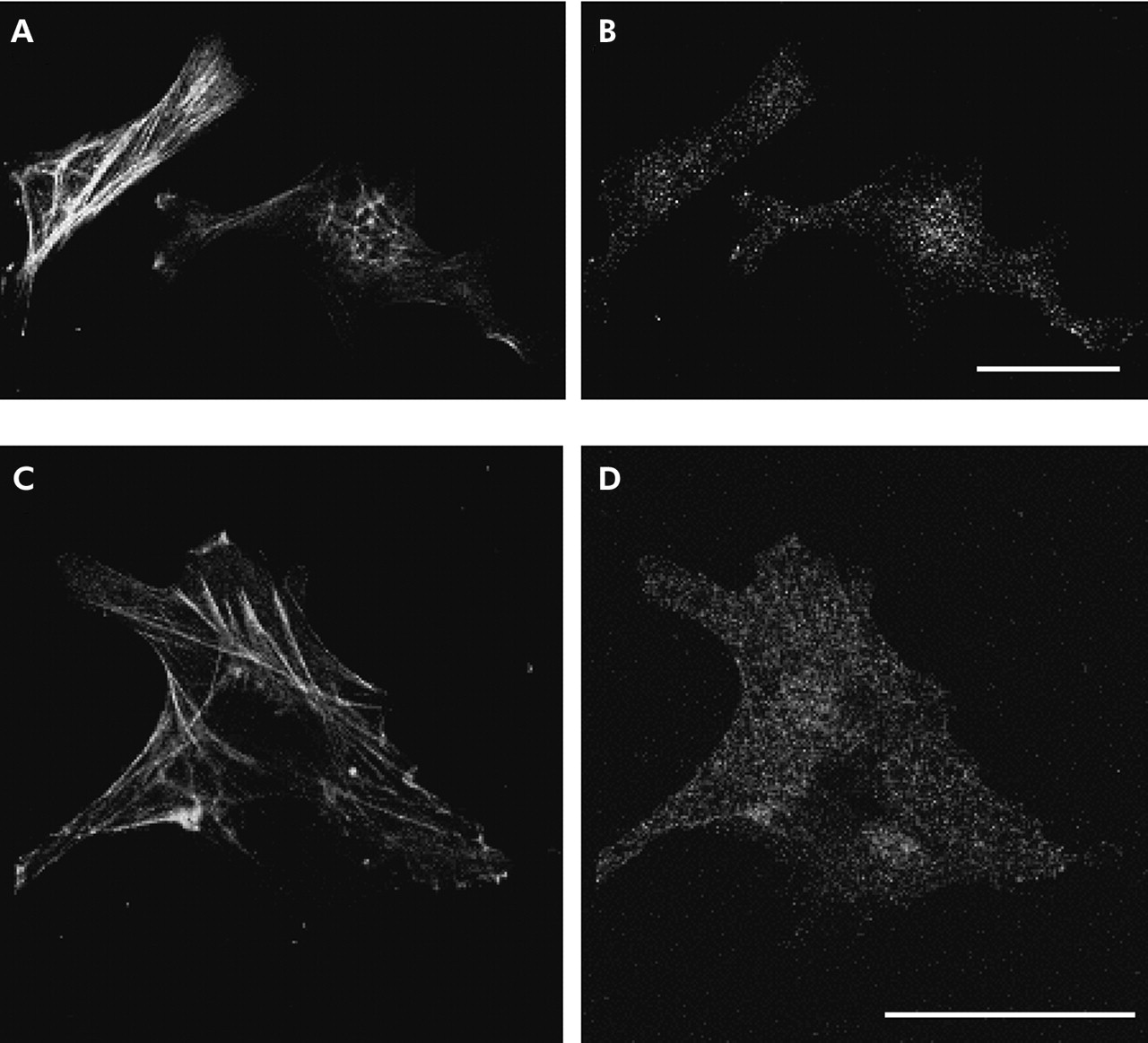
Fluorescence micrographs showing the immunoreactivity of activin A in pancreatic stellate cells (PSCs). Cells were double stained with an anti-activin A rabbit polyclonal antibody (B, D) and an anti-α smooth muscle actin (α-SMA) mouse monoclonal antibody (A, C). Activin A immunoreactivity was observed in PSCs identified with their α-SMA fine network architecture. Bars 40 µm.
Activin A and TGF-β enhanced each other’s secretion and mRNA expression of PSCs
In PSC activation, TGF-β is also known to play a stimulatory role in an autocrine manner.16 To elucidate the interaction of the stimulatory effects of autocrined activin A and TGF-β on PSC activation, we examined their effects on each other’s secretion and mRNA expression of PSCs. As shown in fig 6, activin A enhanced TGF-β secretion from PSCs in a dose dependent manner (fig 6B). In the same manner, TGF-β also increased activin A secretion from PSCs (fig 6A). These data indicate that activin A and TGF-β potentiated each other’s secretion from PSCs. Moreover, activin A and TGF-β increased each other’s mRNA expression in PSCs in a dose dependent manner (fig 6C, D). These data indicate that activin A and TGF-β coordinately activate PSCs. Moreover, these results provide a hypothesis that blockade of autocrined activin A may be able to decrease TGF-β mRNA expression and TGF-β secretion of PSCs, and consequently reduce PSC activation and collagen secretion by blocking both activin A and TGF-β stimuli.

Effect of activin A and transforming growth factor β (TGF-β) on each other’s secretion and mRNA expression of pancreatic stellate cells (PSCs). (A, B) Concentration of activin A (A) and TGF-β (B) secreted from PSCs into culture medium was quantified after 48 hours of incubation with the indicated amounts of TGF-β (A) or activin A (B). Results are means (SEM) of three independent experiments (*p<0.05, **p<0.01). (C, D) mRNA expression of activin A βA subunit (C) and TGF-β (D) was determined by reverse transcription-polymerase chain reaction after 24 hours of incubation with the indicated amounts of TGF-β (C) or activin A (D), using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA expression as a control.
Follistatin decreased TGF-β secretion and mRNA expression of PSCs
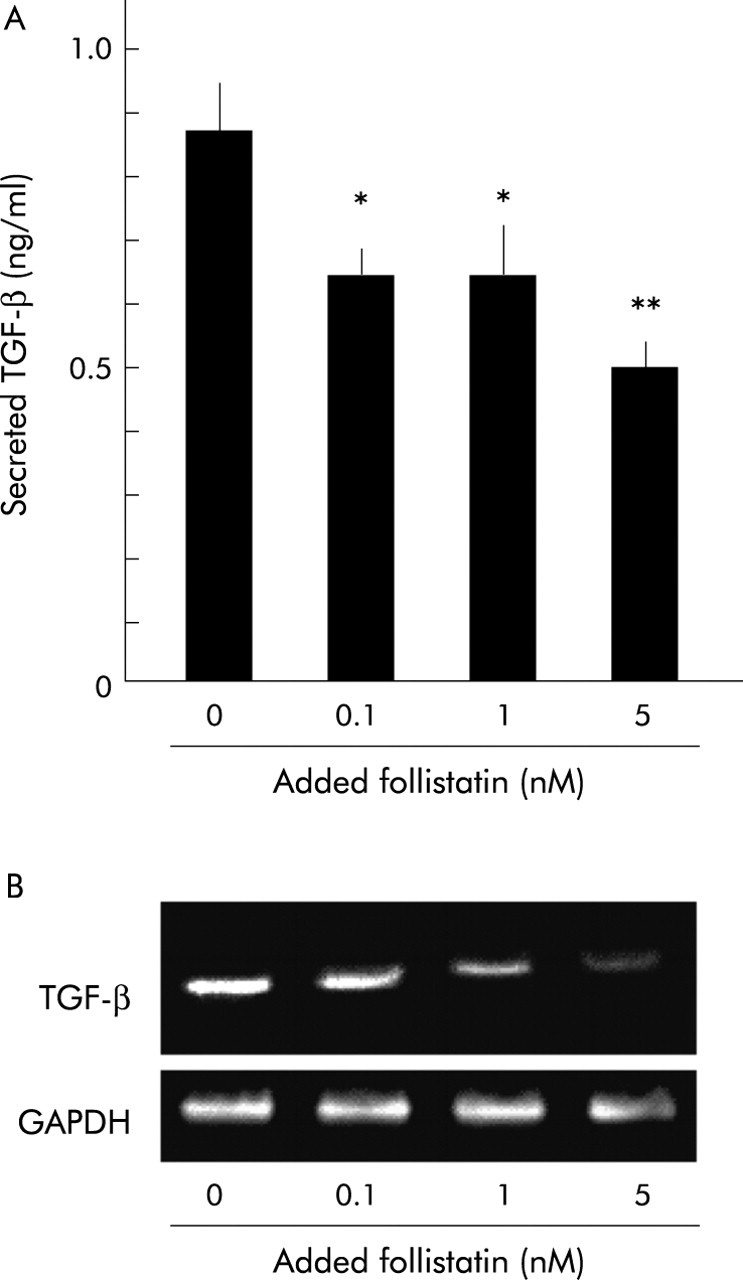
To examine this hypothesis, we investigated the effect of follistatin, an activin A binding protein that blocks activin A activity, on TGF-β secretion and mRNA expression of PSCs. As shown in fig 7, follistatin decreased TGF-β secretion from PSCs in a dose dependent manner (fig 7A). Follistatin also reduced TGF-β mRNA expression in PSCs (fig 7B). Together with the data shown above that activin A enhanced TGF-β secretion from PSCs, it is suggested that follistatin inhibited TGF-β secretion and mRNA expression of PSCs by inhibiting the autocrined activin A stimulus, indicating that follistatin can concurrently inhibit both autocrined activin A and TGF-β stimuli.

Effect of follistatin on transforming growth factor β (TGF-β) secretion and mRNA expression of pancreatic stellate cells (PSCs). (A) Concentration of TGF-β secreted from PSCs into culture medium was quantified after 48 hours of incubation with the indicated amounts of follistatin. Results are means (SEM) of three independent experiments (*p<0.05, **p<0.01). (B) mRNA expression of TGF-β in PSCs was determined by reverse transcription-polymerase chain reaction after 24 hours of incubation with the indicated amounts of follistatin, using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA expression as a control.
Follistatin inhibited PSC activation and collagen secretion
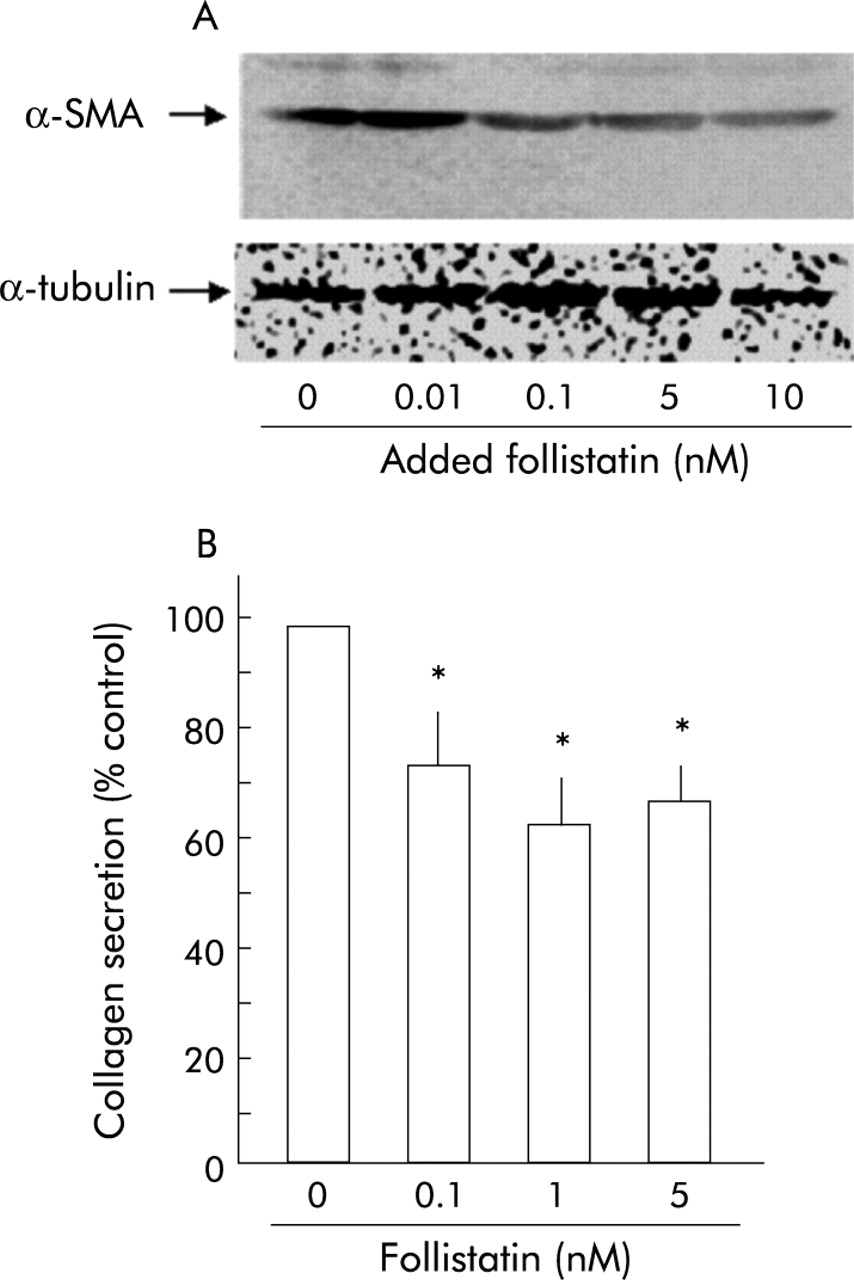
Knowing that follistatin blocks both activin A and TGF-β stimuli to PSCs, we finally examined whether follistatin inhibits PSC activation and collagen secretion. As shown in fig 8A, follistatin decreased α-SMA content in PSCs in a dose dependent manner, indicating that follistatin inhibited PSC autoactivation. In addition, as shown in fig 8B, follistatin inhibited collagen secretion by PSCs. These data indicate that follistatin is a potent inhibitor of PSC activation and collagen secretion.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of follistatin on pancreatic stellate cell (PSC) activation and collagen secretion. (A) Top panel: western blotting of α smooth muscle actin (α-SMA) in total homogenates of PSCs was carried out after 48 hours of incubation with the indicated amounts of follistatin. Follistatin reduced α-SMA expression of PSCs. Bottom panel: the blots were reprobed with anti-α-tubulin antibody. Expression of α-tubulin protein, examined as an internal control, was not altered by activin A. (B) Collagen secretion by PSCs into culture medium during 48 hours of incubation with the indicated amounts of follistatin was quantified with Sirius red dye. Results are expressed as per cent collagen concentration of control. Values are means (SEM) of three independent experiments (*p<0.05).
DISCUSSION
In this study, we demonstrated that activin A activates PSCs and promotes collagen secretion in an autocrine manner. We also showed that activin A and TGF-β enhance each other’s secretion and mRNA expression of PSCs. We also found that follistatin, an activin A binding and blocking protein, inhibits TGF-β secretion and mRNA expression of PSCs and attenuates PSC activation and collagen secretion. These data suggest the participation of activin A in pancreatic fibrosis and the potential application of follistatin in its treatment.
With regard to the effects of activin A on the pancreas, much attention has been directed towards its physiological role in regulation and differentiation of pancreatic islet cells. Activin A has been shown to be present in progenitor cells of fetal pancreas23 and in islet A and D cells of adult pancreatic islets.8 Both in vitro and in vivo studies have revealed that activin A regulates differentiation of pancreatic endocrine cells.9,18 It is also reported that activin A stimulates insulin release from islet B cells.8 To date, however, activin A participation in pancreatic disorders, including pancreatic fibrosis, has been uncertain and its presence in PSCs has not been described. Thus the current study provides the first evidence of activin A participation in pancreatic fibrosis.
The mechanism of digestive organ fibrosis has been extensively studied on hepatic fibrosis, including hepatic stellate cell (HSC) participation. Thus the mechanism of pancreatic fibrosis has been assessed in the analogy of that of hepatic fibrosis. Although activin A has been demonstrated to contribute to hepatic fibrosis and to increase HSC collagen expression,24,25 it is noteworthy that the cellular source of activin A is quite different between the pancreas and liver. In both normal and fibrotic livers, activin A is mainly expressed in parenchymal hepatocytes.7,24 Although HSCs express activin A,26 the cellular population of parenchymal hepatocytes is much greater than that of HSCs in the liver. Thus it is assumed that activin A promotes hepatic fibrosis mainly in a paracrine manner. In contrast, pancreatic acinar cells, a major inhabitant of the pancreas, do not express activin A.8 Although A and D cells in pancreatic islets express activin A, their population is small, especially in fibrotic pancreas. Therefore, it can be postulated that activin A activates PSCs and promotes pancreatic fibrosis in an autocrine manner. Taken together, we suggest that distinct mechanisms underlie pancreatic and hepatic fibrosis.
Our observations that activin A and TGF-β increased each other’s mRNA expression and secretion of PSCs are unique. They imply the existence of an autocrine loop of activin A and TGF-β in PSCs. In parenchymal hepatocytes, both activin A and TGF-β are expressed and regulate their proliferation in an autocrine manner.7,26 However, the interaction between autocrined activin A and TGF-β in hepatocyte growth control is still unknown. The current study demonstrated, for the first time, the interaction between autocrined activin A and TGF-β, suggesting activin A and TGF-β synergistically promote pancreatic fibrosis. Although this interaction implies that both activin A and TGF-β are potent fibrogenic factors, it provides a novel therapeutic strategy for pancreatic fibrosis. Accordingly, blockade of either activin A or TGF-β can inhibit both effects of activin A and TGF-β on PSC activation. Indeed, follistatin reduced TGF-β secretion and mRNA expression of PSCs and potently inhibited PSC activation and collagen secretion (figs 7, 8). These results suggest that follistatin may be a potential therapeutic agent to inhibit pancreatic fibrosis. In this respect, it has been reported that both intraportally and intravenously administrated follistatin accelerated liver regeneration after partial hepatectomy,27,28 suggesting follistatin can play a therapeutic role in vivo. Thus an in vivo study of the inhibitory effects of follistatin on pancreatic fibrosis is warranted.
Although follistatin is a potent inhibitor of TGF-β expression and collagen secretion of PSCs, even high doses of follistatin could not cause complete loss of TGF-β expression or of collagen secretion (figs 7, 8). Recently, Shek et al reported that blockade of autocrined TGF-β by its neutralising antibodies could not completely abolish collagen synthesis by PSCs, which is consistent with our observations.29 These data suggest the involvement of additional mechanisms in the regulation of TGF-β and collagen expression in PSCs apart from the autocrine loop of activin A and TGF-β. In this respect, Mews et al reported that collagen synthesis of PSCs is enhanced by various cytokines such as tumour necrosis factor α and interleukin 10.30 In other cell types, TGF-β expression is increased by interleukin 131 and platelet derived growth factor,32 to which PSCs respond.30 Although it is still uncertain that these cytokines are secreted from PSCs, HSCs have been shown to express and secrete a variety of cytokines, including interleukins.33–35 Thus the possibility that autocrined cytokines other than activin A and TGF-β might regulate collagen and TGF-β expression in PSCs deserves further investigation.
In addition to the effect on cellular differentiation and activation, activin A possesses inhibitory effect on the proliferation of various cells, including pancreatic acinar cells.36 However, exogenous activin A (1–10 nM) did not alter PSC 3H-thymidine uptake. In addition, follistatin (1–10 nM) increased it by only 10–15% (our unpublished data). Thus the inhibitory effect of activin A on PSC proliferation may be, if any, much smaller than that of TGF-β.16
In conclusion, we have shown that activin A is an autocrine activator of PSCs and follistatin inhibited PSC activation. These observations provide new insights for understanding the mechanism of pancreatic fibrosis and developing novel therapeutic strategies.
Acknowledgments
We are grateful to Dr Yuzuru Eto for providing activin A and antiactivin A antibody, and to Ms Mie Shikata for technical and secretarial assistance. This work was supported by grants-in-aid from the ministry of Education, Culture, Sports, Science, and Technology.
REFERENCES
Footnotes
-
↵* N Ohnishi and T Miyata contributed equally to this study.