


Abstract
Signal transduction through the p38 mitogen-activated protein (MAP) kinase pathway is central to the transcriptional and translational control of cytokine and inflammatory mediator production. p38 MAP kinase inhibition hence constitutes a promising therapeutic strategy for treatment of chronic inflammatory diseases, based upon its potential to inhibit key pathways driving the inflammatory and destructive processes in these debilitating diseases. The present study describes the pharmacological properties of the N-phenyl pyridinone p38 MAP kinase inhibitor benzamide [3- [3-bromo-4-[(2,4-difluorophenyl)methoxy]-6-methyl-2- oxo-1(2H)-pyridinyl]-N,4-dimethyl-, (−)-(9CI); PH-797804]. PH-797804 is an ATP-competitive, readily reversible inhibitor of the α isoform of human p38 MAP kinase, exhibiting a Ki = 5.8 nM. In human monocyte and synovial fibroblast cell systems, PH-797804 blocks inflammation-induced production of cytokines and proinflammatory mediators, such as prostaglandin E2, at concentrations that parallel inhibition of cell-associated p38 MAP kinase. After oral dosing, PH-797804 effectively inhibits acute inflammatory responses induced by systemically administered endotoxin in both rat and cynomolgus monkeys. Furthermore, PH-797804 demonstrates robust anti-inflammatory activity in chronic disease models, significantly reducing both joint inflammation and associated bone loss in streptococcal cell wall-induced arthritis in rats and mouse collagen-induced arthritis. Finally, PH-797804 reduced tumor necrosis factor-α and interleukin-6 production in clinical studies after endotoxin administration in a dose-dependent manner, paralleling inhibition of the target enzyme. Low-nanomolar biochemical enzyme inhibition potency correlated with p38 MAP kinase inhibition in human cells and in vivo studies. In addition, a direct correspondence between p38 MAP kinase inhibition and anti-inflammatory activity was observed with PH-797804, thus providing confidence in dose projections for further human studies in chronic inflammatory disease.
Rheumatoid arthritis (RA) is an aggressive autoimmune disease involving complex interactions among T cells, macrophages, synoviocytes, and other immune cells (Firestein, 2003). Analysis of synovial fluid and tissue from RA patients implicated key cytokines, including TNF-α, IL-1β, and IL-6 in the pathogenesis of the disease (Firestein et al., 1990). Further studies shed light on the complex networks within which these cytokines function and the delicate balance between a pro- or an anti-inflammatory outcome (McInnes and Schett, 2007). TNF-α, IL-1β, and IL-6 are macrophage- and fibroblast-derived proteins that induce expression of inflammatory mediators such as COX-2, inducible nitric-oxide synthase, adhesion molecules, and metalloproteinases, resulting in synovial inflammation and associated cartilage and bone destruction. Evidence generated in both animal models and in human studies support the role of TNF-α, IL-1, and IL-6 in the pathogenesis of RA (Dayer et al., 2001; Scott and Kingsley, 2006; Hennigan and Kavanaugh, 2008). Treatment with Remicade, Humira, Enbrel, Kineret, and Tocilizumab reduce joint pain and swelling, and retard progression of bone loss in patients whose disease is unsatisfactorily controlled by conventional disease-modifying antirheumatic drugs (Okamoto et al., 2008). However, these biologic agents are limited due to the requirement for parenteral administration, difficulty in dose titration, poor reversibility, induction of host neutralizing antibody response, high production costs, and a significant population of patient refractory to the treatment.
An alternative approach for the modulation of RA critical proinflammatory cytokines is the manipulation of the signaling cascades involved in their production. It is interesting to note that a marked overexpression of the phosphorylated (activated) form of p38 MAP kinase was demonstrated in RA synovium (Schett et al., 2000). p38 kinase along with ERK and JNK are key MAP kinase signaling enzymes that cells use to adapt to inflammatory and stressful conditions. Pathogens or inflammatory stimuli initiate a phosphorylation cascade mediated by p38 kinase that leads to the transcription and translation of inflammatory response-associated genes that encode proteins such as TNF-α, IL-1, IL-6, and IL-8. Activation of the p38 pathways also leads to the production of anti-inflammatory cytokines such as IL-10 (Chanteux et al., 2007), activation of negative feedback loops initiated by the phosphorylation of transforming growth factor β-activated kinase-1-binding protein (Shin et al., 2009) as well as up-regulation of MAP kinase phosphatases that dephosphorylate and inactivate p38 kinase, ERK, and JNK (Wadgaonkar et al., 2004). These multiple positive and negative pathway components highlight the complex feedback and feedforward aspects of the p38 signaling cascade (Hu et al., 2008).
The p38 MAP kinase family consists of four isoforms: α, β, γ, and δ. These kinases show a high degree of sequence homology, with 60 to 75% overall sequence identity and >90% within the kinase domain (Kumar et al., 1997). The α and β isoforms are ubiquitously expressed, with the α isoform being the better characterized in terms of its role in inflammation. Although studies using a chemical genetic approach in mice indicate that the β isoform does not play a role in the regulation of inflammation (O'Keefe et al., 2007), it may have a role in pain (Svensson et al., 2005). The γ and δ isoforms are expressed in a tissue-restricted manner, with γ expressed in skeletal muscle (Li et al., 1996) and δ expressed in lungs, kidneys, testis, pancreas, and intestines (Kumar et al., 1997).
Due to the apparent role of p38α kinase in inflammation, its therapeutic potential has been extensively studied. Several small-molecule inhibitors of p38 kinase have been generated and characterized. These include VX-745, VX-702, SCIO-323, SCIO-469, AMG-548, BIRB 796, and pamapimod (Pettus and Wurz, 2008; Cohen et al., 2009). A substantial number of these inhibitors have progressed into human clinical studies, and development was discontinued due to unacceptable safety profiles (Pettus and Wurz, 2008). Common side effects include skin rash, elevated liver enzymes, and gastrointestinal disorders. Because these adverse effects vary with chemotype, and each of these compounds have distinct kinase selectivity patterns, the toxicities observed may be structure- rather than mechanism-based.
PH-797804 is a highly selective inhibitor of p38α kinase. In cell-based assays as well as animal models of acute inflammation, PH-797804 blocked the production of cytokines and proinflammatory mediators. Furthermore, PH-797804 demonstrated robust anti-inflammatory activity in chronic disease models, significantly reducing both joint inflammation and associated bone loss in streptococcal cell wall-induced arthritis in rats and mouse collagen-induced arthritis. Results from an endotoxin challenge model in humans demonstrated the ability of PH-797804 to modulate lipopolysaccharide (LPS)-stimulated cytokine production in a dose-dependent and concentration-dependent manner.
Materials and Methods
Preparation of PH-797804
PH-797804 was prepared by the Pfizer Discovery Medicinal Chemistry Department (St. Louis, MO).
In Vitro Assays
Kinase Activation.
The activation of p38α kinase by mitogen-activated protein kinase kinase (MKK) 6 was carried out in the presence of 300 μM ATP, 10 mM magnesium acetate, and 25 mM HEPES, pH 7.5. Constitutively active MKK6 was used for activation of p38α kinase at a molar ratio for p38α:MKK6 ranging from 50:1 to 100:1. The reaction mixture was allowed to incubate at 30°C for 1 h. The activation of JNK2 by MKK7 was carried out at 30°C for 1 h in the presence of 500 μM ATP, 10 mM magnesium acetate, and 25 mM HEPES, pH 7.5. The constitutively active form of MKK7 was added to yield a 50:1 M ratio of JNK2/MKK7. The enzyme was either used immediately or measured (in aliquots) and stored at −80°C.
Kinase Activity Assay.
A resin capture assay method was used to determine the phosphorylation of epidermal growth factor receptor peptide (EGFRP) or GST-c-Jun by p38 kinases or JNK2, respectively. Reactions mixtures contained 25 mM HEPES, pH 7.5, 10 mM magnesium acetate, ATP (at the indicated concentration), 0.05 to 0.3 μCi of [γ-33P]ATP, 0.8 mM dithiothreitol, and either 200 μM EGFRP or 10 μM GST-c-Jun for p38α kinase or JNK2 reactions, respectively. The reaction was initiated by the addition of either 25 nM p38α kinase or 100 nM JNK2 to give a final volume of 50 μl. The JNK2 and p38α kinase reactions were incubated at 25°C for either 20 or 30 min, respectively. Under these conditions, the formation of product for both p38α kinase and JNK2 was linear with time. The reaction was stopped, and the unreacted [γ-33P]ATP was removed by the addition of 150 μl of AG 1 × 8 ion exchange resin in 900 mM sodium formate, pH 3.0. Once thoroughly mixed, solutions were allowed to stand for 5 min. A 50-μl aliquot of head volume containing the phosphorylated substrate was removed from the mixture and transferred to a 96-well plate. MicroScint-40 scintillation cocktail (150 μl; PerkinElmer Life and Analytical Science, Boston, MA) was added to each well and the radioactivity quantitated using a TopCount NXT microplate scintillation and luminescence counter (PerkinElmer Life and Analytical Science, Boston, MA).
Inhibition Studies.
The initial velocities in the presence and absence of PH-797804 were obtained using ATP/[γ-33P]ATP as the varied substrate. The peptide (EGFRP) or protein substrate (GST-c-Jun) was fixed at a single concentration. The initial velocities were determined in triplicate using the assay described above. The inhibition data were fit to the competitive inhibition (eq. 1), noncompetitive inhibition (eq. 2), or uncompetitive inhibition (eq. 3) by using GraFit version 4.0 service pack (Erithacus Software, Surrey, UK).


 In these equations, Vmax is the maximum velocity, Km is the Michaelis-Menten constant for the varied substrate (ATP), S is the concentration of the varied substrate, I is the concentration of the inhibitor, and Kis and Kii are the dissociation constants for the interaction of inhibitor with the free enzyme or the substrate-bound enzyme, respectively.
In these equations, Vmax is the maximum velocity, Km is the Michaelis-Menten constant for the varied substrate (ATP), S is the concentration of the varied substrate, I is the concentration of the inhibitor, and Kis and Kii are the dissociation constants for the interaction of inhibitor with the free enzyme or the substrate-bound enzyme, respectively.
Ligand Exchange Reaction.
The competition between inhibitors and a fluorescent probe for binding to nonphosphorylated, nonactive p38α kinase was monitored in real-time by using stopped flow instrumentation (TgK Scientific Limited, Bradford-on-Avon, UK) equipped with a fluorescence detector. The fluorescence resonance energy transfer signal from probe binding to nonactive p38α kinase was measured by exciting the sample at 295 nm and monitoring the fluorescence intensity using a 420 nm cut-off filter. The buffer used for all ligand exchange reactions was 25 mM HEPES, pH 7.5, with 10% dimethyl sulfoxide. In the first ligand exchange reaction, 100 nM nonactive p38α kinase is equilibrated with 250 nM probe (Kd = 13 nM) and then diluted into solution containing the unlabeled inhibitor (at time 0). In the second reaction, 100 nM nonactive p38α kinase is equilibrated with the unlabeled inhibitor and then diluted into solution containing the probe (at time 0). The final concentrations of reactants are held constant so that both reactions have the same response at equilibrium (Morelock et al., 1995).
Progress Curve Analysis.
The binding constants of PH-797804 were determined using a competition assay as described previously (Pargellis et al., 2002). The binding constants were calculated based on a one-step binding model using the numerical integration software DYNAFIT (Kuzmic, 1996).
Cell-Based Assays
Cell Lines and Primary Human Monocytes.
The U937 human premonocytic cell line was obtained from the American Type Culture Collection (Manassas, VA). These cells were differentiated to a monocytic/macrophage phenotype as described by Burnette et al. (2009). Rheumatoid arthritis synovial fibroblasts (RASF) were derived from the inflamed synovium of a female RA patient who was undergoing total knee replacement. Synovial tissue was teased away from adjacent cartilage and dispersed into single cells with collagenase. Cells were expanded and banked. RASF were further cultured as described by Burnette et al. (2009). Primary human monocytes were obtained from venous blood of donors collected anonymously with informed consent at an on-site clinic into sodium heparin tubes. Monocytes were isolated as described previously (Burnette et al., 2009).
Stimuli-Induced MAP Kinase Activation.
Cells (differentiated U937 cells) pretreated with or without inhibitors for 1 h, were stimulated with LPS (0.1 μg/ml) for 30 min, washed twice with ice-cold phosphate-buffered saline, and lysed with 150 mM NaCl, 20 mM Tris, pH 7.5, 1% Triton X-100, 5 mM EDTA, 50 mM NaF, 10% glycerol, and 1 mM Na3VO4 and protease inhibitors (Boehringer Mannheim, Indianapolis, IN) for immunoprecipitation in vitro kinase assays.
Immunoprecipitation in Vitro Kinase Assay.
The protein content of the U937 cell lysates was determined by Bradford assay (Bradford, 1976). Lysates (750 μg) were immunoprecipitated with anti-MK-2 antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and protein A/G-agarose (2–3 h; 4°C). Immune complexes were washed three times with lysis buffer, once with kinase assay buffer (50 mM HEPES, 10 mM MgCl2, 5 mM MnCl2, and 1 mM dithiothreitol), and resuspended in kinase assay buffer with 4 μg of HSP27 substrate and 0.5 μCi of [γ-33P]ATP. After incubation at 30°C for 15 min, the reaction was stopped by adding an equal volume of 2× SDS-sample buffer, and heated for 3 min. Reaction mixtures were resolved by 4 to 20% SDS-polyacrylamide gel electrophoresis, and radiolabeled substrate was visualized by autoradiography. The labeled bands were excised from the gel, scintillation cocktail was added, and the bands were counted on an LS6000IC counter (Beckman Coulter, Fullerton, CA). MK-2 is selectively activated by p38 kinase in U937 cells, and its phosphorylation of HSP27 is thereby an indirect measurement of p38 kinase activity.
LPS-Induced Cytokine (TNF-α and IL-1β) Production.
Differentiated U937 cells or purified human monocytes were seeded into 96-well tissue culture plates (200,000 cells/well) in complete media. After 24 h, the cells were pretreated for 60 min in the presence or absence of PH-797804 and then stimulated with LPS (0.1 μg/ml) for 4 h (U937 cells) or 16 h (monocytes). Culture media were then collected for determination of TNF-α or IL-1β levels by Luminex bead array technology (Linco/Millipore, Billerica, MA). Cytokine concentrations were extrapolated from recombinant protein standard curves using a BioAssay Solver macro with a four-parameter logistic model and solving for IC50 after iterating to the best least-squares fit.
LPS-Induced TNF-α and IL-1β Production in Human Whole Blood.
Venous blood was collected with informed consent as described above. Assay of LPS-induced cytokine production was performed as described by Burnette et al. (2009) with the exception that the final LPS concentration was 10 μg/ml, the incubation period for IL-1β production was 24 h, and cytokine analysis was done using the Luminex bead array technology (Linco/Millipore).
IL-1β-Induced PGE2 Production.
RASF were seeded into 96-well tissue culture plates (5 × 104 cells/well) in complete growth medium. After 24 h, the medium was replaced with fresh growth medium containing 1% fetal bovine serum. Cells were treated with serial concentrations (10,000–0.01 nM) of PH-797804 or dimethyl sulfoxide vehicle control for 1 h, stimulated with 1 ng/ml IL-1β (R&D Systems, Minneapolis, MN) for 18 to 20 h at 37°C, and then conditioned media were collected. PGE2 levels the in cultured media were quantitated by ELISA (Cayman Chemical, Ann Arbor, MI).
Osteoclast Formation from Rat Bone Marrow Cells.
Male Lewis rats weighing 250 to 300 g were obtained from Harlan Industries (Houston, TX). Rat bone marrow cells were collected from femora and tibiae of the animals. Cells were washed twice with serum-free α-minimal essential medium (Invitrogen, Carlsbad, CA) containing 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen). Cells were plated at a density of 1.7 × 106/cm2 in α-minimal essential medium supplemented with 10% (v/v) fetal bovine serum (Invitrogen), incubated at 37°C, in an atmosphere of 95% air and 5% CO2, and then treated with a combination of recombinant mouse RANKL (100 ng/ml) and recombinant mouse macrophage–colony-stimulating factor (M-CSF) (10 ng/ml) (R&D Systems) along with PH-797804 at various concentrations (0.0001–1 μM). The cytokine concentrations used had been shown to induce maximal osteoclast formation in previous studies (data not shown). After 2 days of culture, half of the culture medium was gently removed, to minimize loss of nonadherent cells, and replaced with an equal volume of fresh culture media containing cytokines and p38 kinase inhibitors. On day 3, the medium was removed, and the cells were fixed by the addition of a 60% (v/v) acetone solution prepared in sodium citrate buffer, pH 5.4, for 30 s. The fixed cells were washed twice with distilled water and air-dried. Tartrate-resistant acid phosphatase (TRAP)-positive (TRAP+) cells were detected using a TRAP staining kit (Sigma-Aldrich, St. Louis, MO). Osteoclasts, defined as cells TRAP+ multinucleated cells with three or more nuclei were counted under a light microscope by using computer-assisted AnalySiS Pro software (Soft Imaging System, Münster, Germany). The IC50 value determinations were made using a four-parameter logistic analysis.
Cell Viability Assay.
Cell viability was determined as described previously (Burnette et al., 2009).
In Vivo Studies
Animal Use.
The Pfizer Institutional Animal Care and Use Committee reviewed and approved the animal use in these studies. The animal care and use program is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International. Adult male Lewis rats (225–250 g; Harlan, Indianapolis, IN) or adult male cynomolgus monkeys (6–8 kg; Charles River Laboratories, Houston, TX) were used in the LPS-induced TNFα production studies. Female Lewis rats (125–140 g; Harlan) were used in the SCW arthritis model and DBA/1 mice (8–12 weeks old; The Jackson Laboratory, Bar Harbor, ME) were used in the collagen-induced arthritis (CIA) model.
LPS-Induced TNF-α Production in Rats.
Rats were fasted 18 h before oral dosing and were allowed free access to water throughout the experiment. Each treatment group consisted of five animals. PH-797804 was prepared as a suspension in a vehicle consisting of 0.5% methylcellulose (Sigma-Aldrich) and 0.025% Tween 20 (Sigma-Aldrich). The compound or vehicle was administered by oral gavage in a volume of 1 ml. Two vehicle groups were used per experiment to control for intraexperiment variability, and two experiments were performed. LPS (Escherichia coli serotype 0111:B4; Sigma-Aldrich) was administered 4 h after compound intravenous injection at a dose of 1 mg/kg in 0.5 ml of sterile saline (Baxter Healthcare, Deerfield, IL), a dose determined previously to be optimal (data not shown). Blood was collected in serum separator tubes via cardiac puncture 90 min after LPS injection, a time point corresponding to maximal TNF-α production (data not shown). After clotting, serum was withdrawn and stored at −20°C until it was assayed for TNF-α by ELISA (Burnette et al., 2009).
LPS-Induced TNF-α Production in Cynomolgus Monkeys.
The study was conducted at Charles River Laboratories in accordance with the institutional guidelines for humane treatment of animals. Adult male cynomolgus monkeys were fasted with free access to water for at least 18 h before testing. TNF-α was induced in cynomolgus monkeys (n = 3) by a single intravenous aqueous injection of LPS (E. coli serotype 055:B5) (Sigma-Aldrich) at a dose of 10 μg/kg in a volume of 0.5 ml of sterile saline. This dose of LPS had been shown previously to produce nanogram per milliliter levels of TNF-α in rats (data not shown). PH-797804 was delivered through a nasogastric tube in a 1-ml volume 2 h before LPS in an aqueous vehicle of 0.5% methylcellulose (w/v) and 0.1% Tween 80 (v/v). Blood was collected into lithium heparin Vacutainer tubes just before LPS injection and 0.5, 0.75, 1, 1.25, and 2 h after LPS injection. Plasma TNF-α was determined using a human TNF-α ELISA kit (BD Biosciences Pharmingen, San Diego, CA) that detected human TNF-α with a sensitivity of 7.5 pg/ml. A crossover design was used whereby each of three monkeys received 0.03, 0.1, 0.3, and 1.0 mg/kg PH-797804 with 3-week intervals between treatments. Vehicle response for each monkey was determined as the mean LPS-stimulated TNF-α levels from four nondrug studies. The four nondrug studies were done 2 to 9 months before the PH-797804 studies, with a minimum interval of 3 weeks between studies. The PH-797804 dose, maximal plasma concentration (Cmax), and are under the curve0–24h required to produce 50 and 80% inhibition of peak TNF-α levels were determined using a four-parameter logistic model with two parameters fixed to 0% for minimum and 100% for maximum.
Streptococcal Cell Wall-Induced Arthritis in Rats.
Arthritis was induced in female Lewis rats by a single intraperitoneal administration of peptidoglycan-polysaccharide complexes isolated from group A SCW (15 μg/g body weight). The SCW preparation was purchased from Lee Laboratories (Grayson, GA). The disease course is biphasic in which an acute inflammatory arthritis develops within days 1–3 (non–T-cell-dependent phase) followed by a chronic erosive arthritis (T-cell-dependent phase) developing on days 14 to 28 (Kuiper et al., 1998). Only animals developing the acute phase were treated with PH-797804 from days 10 to 21 after SCW injection. Paw volume was measured on day 21 by using a water displacement plethysmometer. PH-797804 was prepared as an aqueous suspension in 0.5% methylcellulose and 0.025% Tween 20 (Sigma-Aldrich). The compound was administered by oral gavage in a volume of 0.5 ml beginning on day 10 post-SCW injection and continuing daily until day 21. Animals were dosed between 0.015 and 5 mg/kg b.i.d. with PH-797804. Methylcellulose/Tween 20 vehicle was used for comparison. Group size was four to eight animals per group. Two paw volumes were taken for each animal. Paw volume was measured on day 21 by using a water displacement plethysmometer. Three to four paws from each treatment group were scanned for bone density evaluation. Plasma samples were collected on day 21 for determination of compound levels.
Bone Density Determination.
Hind paws obtained from (day 21) arthritic rats were submitted for bone density analysis. Paws were stored at −80°C until use. Bone density was determined using Lunar PixiMus densitometer (GE Medical Systems, Madison, WI) (Nagy and Clair, 2000). Bone density was evaluated in hind paws from the heel through half of the length of the paw and values were expressed as grams per square millimeter.
Collagen-Induced Arthritis in Mice.
Arthritis was induced in DBA/1 mice by injection of 100 μl of 50 μg of chick type II collagen (CII) (provided by Dr. M. Griffiths, University of Utah, Salt Lake City, UT) in Complete Freund's adjuvant (Sigma-Aldrich) at the base of the tail on day 0. The mice were boosted on day 21 as described above. Compound administered in chow was mixed to deliver the approximate dose per day indicated in Fig. 7. The dosing of compound was based on a 20- to 25-g mouse consuming 4 g of chow per day. The mix for 4, 12, 40, and 120 mg/kg/day was 25, 76, 250, and 760 ppm, respectively. The chow was given ad libitum on days 21 through 56. Dose groups consisted of 10 animals per group. Animals were evaluated several times each week for signs of arthritis for up to 8 weeks. Any animal with paw redness or swelling was counted as arthritic. Scoring of severity was carried out using a score of 1 to 3 for each paw (maximal score of 12/mouse). Animals displaying any redness or swelling of digits or the paw were scored as 1, gross swelling of the whole paw or deformity was scored as 2, and ankylosis of joints was scored as 3. Any animal demonstrating a score of 1 or more was considered arthritic. Statistical analysis comparing the percentage of arthritis between groups was done using Fisher's exact test.
Human Endotoxin-Induced Inflammatory Response Model.
This was a single-center, randomized, subject- and investigator-blind/sponsor-open placebo-controlled study. This study was conducted in compliance with the ethical principles derived from the Declaration of Helsinki and in compliance with the Institutional Review Board and informed consent regulations. The study was conducted at the MDS Pharma Services Clinical Center (New Orleans, LA). Included in this study were 36 healthy male subjects from the ages of 18 to 55 years. After an 8-h fast, subjects were administrated a single oral dose of study medication (1, 2, 4, 13, or 30 mg of PH-797804 or placebo) with 300 ml of water (day 1). Twenty-four hours later (day 2), a bolus intravenous injection of 2 ng/kg LPS was administered to all subjects. Based on pharmacokinetic data obtained after administration of single oral doses from 0.3 to 30 mg, it was determined that relatively constant plasma concentrations are present from 24 to 48 h after dosing. Thus, the timing of the LPS administration was chosen to be 24 h after the dose of PH-797804. Blood samples to determine the plasma pharmacokinetics of PH-797804 were collected immediately before study drug administration as well as 24, 29, and 48 h after study drug administration. Plasma PH-797804 concentrations were determined using liquid chromatography-tandem mass spectrometry (LC-MS/MS). The analytical range of the assay was 0.02 to 20 ng/ml. Blood samples for measurements of cytokines in the plasma were collected just before study drug administration, just before LPS administration, and serially up to 5 h after LPS administration. Blood samples for measurement of MK-2 activity in whole blood were collected at just before LPS administration up to 5 h after LPS administration. TNF-α in plasma was assayed by ELISA, with a dynamic range of 15 to 1000 pg/ml. IL-6 in plasma was assayed using Luminex multiplex ELISA technology, with a dynamic range of 3.2 to 10,000 pg/ml. MK-2 activity in whole blood lysates was determined as described by Burnette et al. (2009). In brief, whole blood samples were lysed in a buffer containing protease and phosphatase inhibitors. The lysates were processed using QiaShredder tubes (QIAGEN, Valencia, CA), and the resulting supernatants were used for the assay. The assay was a radiometric assay consisting of capture of MK-2, in vitro phosphorylation of biotin-tagged HSP27 peptide, and quantitation of the radiolabeled phospho-HSP27 by capture on a streptavidin-coated flashplate and scintillation counting (Burnette et al., 2009).
Measurement of PH-797804 in Rat and Monkey Plasma.
PH-797804 concentrations in rat and monkey plasma were determined using an extraction procedure followed by LC-MS/MS. PH-797804 standards were prepared by spiking a stock solution in dimethyl sulfoxide (Mallinckrodt Baker Inc., Phillipsburg, NJ) into normal plasma and performing serial dilutions to achieve a nine-point calibration curve. For rat, 20 to 40 μl of plasma was extracted by adding acetonitrile (Honeywell Burdick & Jackson, Muskegon, MI) containing analytical internal standard to a final volume of 200 μl, and the samples were vortex-mixed and centrifuged to precipitate proteins. For monkey, 50 μl of plasma was combined with acetonitrile containing analytical internal standard to a final volume of 200 μl, and the samples were extracted using solid phase extraction. Monkey plasma samples were applied to an Oasis HLB solid-phase extraction 96-well plate (Waters, Milford, MA) that had been preactivated with 100% methanol followed by 2% acetic acid in water and then washed with two 400-μl aliquots of methanol/2% acetic acid in water [30:70 (v/v)] followed by two 150-μl elutions of the analytes with 100% methanol. The combined eluate was dried down under nitrogen, reconstituted in 100 μl of acetonitrile/0.1% formic acid in water [25:75 (v/v)], and vortex-mixed. A volume of each extract was injected into the LC-MS/MS system using a Chiracel OJ-RH column (Chiral Technologies, Inc., Exton, PA) or Thermo Aquasil C18 column (Thermo Fisher Scientific, Waltham, MA) and a 1% formic acid (Mallinckrodt Baker Inc.) and acetonitrile gradient. An API 4000 Sciex mass spectrometer (Applied Biosystems, Foster City, CA) with turbo-ionspray source was operated in the multiple reaction-monitoring mode to detect PH-797804 and the internal standard in the samples. PH-797804 peak area ratios were determined using the m/z 477→127 transition of PH-797804 versus the m/z transition of the internal standard, and plasma concentrations were calculated using linear regression analysis of the calibration samples. The detection limits of the assay were 0.076 to 5000 ng/ml PH-797804.
Determination of PH-797804 Plasma Protein Binding.
The protein binding of PH-797804 in rat, monkey and human plasma was determined in vitro. Plasma was fortified with PH-797804 to achieve concentrations in the projected therapeutic range and also 10- and 100-fold higher (0.1, 1.0, and 10 μg/ml). Triplicate aliquots of plasma matrix standards underwent equilibrium dialysis against an equal volume of phosphate-buffered saline in an in-house-devised 96-well apparatus similar to that described by Kariv et al. (2001) by using dialysis membranes with a molecular weight cut-off of 12 to 14 kDa. The dialysis was performed in a 37°C shaking incubator for 6 h. The fraction unbound was calculated from the concentrations of PH-797804 in the protein-rich and buffer matrices using the LC-MS/MS method described above. The following equation was used to calculate the fraction bound at the end of the incubation period:
 where Ct is concentration of analyte in plasma after incubation and Cf is concentration of analyte in buffer after incubation.
where Ct is concentration of analyte in plasma after incubation and Cf is concentration of analyte in buffer after incubation.
The following equation was used to calculate the fraction unbound:
 The plasma protein binding of PH-797804, averaged over the range of 0.1 to 10 μg/ml, was 97.8, 93.4, and 96.7% in rat, monkey, and human, respectively, and fraction unbound was 2.2, 6.6, and 3.4%, respectively.
The plasma protein binding of PH-797804, averaged over the range of 0.1 to 10 μg/ml, was 97.8, 93.4, and 96.7% in rat, monkey, and human, respectively, and fraction unbound was 2.2, 6.6, and 3.4%, respectively.
Results
PHA-797804 Is a Potent and Selective Inhibitor of p38α Kinase.
PH-797804 (Fig. 1) is a novel pyridinone inhibitor of the MAP kinase signaling enzyme p38α kinase. Binding, enzyme kinetics, and crystallographic analyses support the ATP-competitive nature of PH-797804 inhibition of p38α kinase activity as well as its high degree of selectivity. PH-797804 inhibited p38α kinase, with an IC50 value of 26 nM (Table 1), and the inhibition is best described by a competitive model as shown in Fig. 2A. Nonlinear fit of the initial velocity data to an equation describing competitive inhibition (see eq. 1 under Materials and Methods) are shown in Fig. 2A. The competitive nature of PH-797804 is apparent in the plot (Fig. 2B), which depicts a family of lines intersecting on the 1/v axis, indicative of competitive inhibition. The average enzymatic competitive inhibition constant (Ki) for PH-797804 toward p38α kinase is 5.8 ± 0.3 nM (Table 1). In contrast, PH-797804 did not inhibit the related MAP kinase JNK2 at concentrations up to 200 μM.

Structure of PH-797804.
PH-797804 affinity and potency for inhibiting recombinant p38 and JNK2 kinases
Data are expressed as the mean value of three independent experiments ± S.E.M.

Competitive inhibition pattern for PH-797804 versus p38α kinase. Initial velocities were obtained with ATP/[γ-33P]ATP as the varied substrate (50, 100, 200, 500, 1000, and 2000 μM). The EGFRP was held constant at 200 μM. The concentrations of PH-797804 were 0 (○), 6 (●), 12 (□), 25 (■), and 50 nM (▵). A, nonlinear fit of data to a competitive inhibition model using GraFit 4.0 service pack. B, double reciprocal plot of the data shown in Fig. 1A. In this experiment, the enzymatic competitive inhibition constant for PH-797804 equals 6.06 ± 0.8 nM (S.E.M.) in this representative experiment.
Ligand exchange reactions were used to determine binding association and dissociation rate constants as indicators of the time dependence and reversibility for PH-797804. The fluorescence resonance energy transfer signal from a fluorescent probe was monitored in the presence of inhibitor to determine the nature of the interaction of PH-797804 with unactivated p38α kinase. PH-797804 demonstrates rapid association and dissociation rates in these experiments. The association rate (Kon) and dissociation rate (Koff) value for PH-797804 is 1.53 × 107 ± 2.88 × 106 M−1 · s−1 and 0.058 ± 0.008 s−1, respectively. In addition, the t1/2 value for dissociation was determined to be 12.6 ± 1.9 s. The binding Ki value determined in these ligand exchange experiments was 3.9 ± 0.3 nM, which is in good agreement with the enzymatic Ki of 5.8 ± 0.3 nM (Table 1). These results suggest that PH-797804 acts as a reversible inhibitor of p38α kinase with fast on and fast off kinetics and suggests that PH-797804 binds to both activated and unactivated p38α kinase with similar affinity.
The profile of PH-797804 for inhibition of other protein kinases and nucleotide binding proteins suggests remarkable selectivity for p38 kinase. PH-797804 inhibited the activity of the closely related p38β kinase isoform, with a Ki value of 40 nM, resulting in a p38α kinase to p38β kinase selectivity ratio of 6.9-fold (Table 1). No significant inhibition of the remaining two human p38 isoforms, p38γ and p38δ kinases, or other MAP kinases, JNK1, JNK2, JNK3, and ERK2, were observed at concentrations of PH-797804 up to 200 μM (Table 2). The selectivity of PH-797804 against the JNK2 kinase (binding Ki ratio >50,000-fold) is significantly greater than that observed for the p38 kinase inhibitor SB203580 (16-fold binding Ki ratio; data not shown). In addition to the kinases noted above, PH-797804 was profiled against approximately 65 kinase and nucleotide binding proteins, and no cross-reactivity was observed (selectivity ratio >500-fold; Table 2) (Xing et al., 2009).
Selectivity profile of PH-797804
PH-797804 Blocks Inflammation-Induced Production of Cytokines and Proinflammatory Mediators in Cellular Assays Relevant to Rheumatoid Arthritis and Inflammation.
PH-797804 was potent and efficacious in blocking cellular p38 kinase activity along with biological responses in human cells associated with inflammation in the arthritic joint. LPS-stimulated TNF-α production and p38 kinase activity as measured by immune complex kinase assay of the downstream kinase MK-2 in the human monocytic U937 cell line were inhibited, with comparable IC50 values of 5.9 and 1.1 nM, respectively (Table 3; Fig. 3A). Although PH-797804 was shown to inhibit cellular p38 kinase activity as assessed by either MK-2 immune complex kinase assay or p38 kinase-dependent phosphorylation of HSP27, no inhibitory effect was observed on either the JNK pathway (c-Jun phosphorylation) or ERK pathway (ERK phosphorylation) in U937 cells at concentrations up to 1 μM (Xing et al., 2009).
Cellular activity of PH-797804 on cytokine production, p38 activity, and PGE2 production—cell types relevant to rheumatoid arthritis and inflammation

PH-797804 inhibition of cytokine, PGE2 production, and p38 MAP kinase activity in human monocytic cells, human whole blood, and rheumatoid arthritis synovial fibroblasts. A, LPS-challenged U937 cells (human monocytic cell line). Data are from a single representative experiment (duplicate determinations for each data point) in which U937 cells were stimulated with LPS for 4 h after a 60-min incubation in the presence or absence of the indicated concentrations of PH-797804. Culture supernatants were collected and assayed for TNF-α by Luminex multiplex TNF-α kits. Fitting the data to a standard curve generated with known amounts of recombinant TNF-α protein yielded an IC50 value of 7.35 nM. The control value (+LPS) for this experiment was 1.96 ng/ml TNF-α for the designated time. Data from three separate experiments yielded an IC50 of 5.9 ± 1.3 nM (mean ± S.E.M.; Table 3). The inhibition of p38 kinase activity was measured indirectly by evaluating the effect of PH-797804 on the ability of a downstream enzyme (MK-2) to phosphorylate its substrate HSP27. U937 cells were stimulated with LPS for 30 min, after a 60-min preincubation in the presence or absence of the indicated concentrations of PH-797804. Cell lysates were prepared, MK-2 was immunoprecipitated, and activity was determined as described under Materials and Methods. Data are from two independent experiments (IC50 = 1.05 ± 0.64; mean ± S.D.). B, IL-1β challenged rheumatoid arthritis synovial fibroblasts. RASF were incubated with PH-797804 for 1 h. IL-1β (1 ng/ml) was added to the assay plate and incubated for 20 h at 37°C with 5% CO2. PGE2 levels in cultured media were determined by ELISA. Data plotted are from a single representative experiment from a total of six experiments each performed in duplicate. C, LPS-challenged isolated human monocytes. Data are from a single representative experiment preformed in duplicate in which purified human monocytes were stimulated with LPS for 16 h after a 60-min incubation in the presence or absence of the indicated concentrations of PH-797804. Culture supernatants were collected and assayed for TNF-α and IL-1β by Luminex multiplex cytokine kits. Fitting the data to a standard curve generated with known amounts of recombinant TNF-α and IL-1β protein yielded IC50 values of 3.0 nM each. Control values (+LPS) for this experiment were 9.4 and 1.76 ng/ml for TNF-α and IL-1β, respectively. Data from three separate experiments yielded IC50 values of 3.4 ± 2.0 and 3.4 ± 0.5 nM for TNF-α and IL-1β, respectively (mean ± S.E.M.) (see Table 3). D, LPS-challenged human whole blood. Human whole blood was incubated with PH-797804 for 1 h. LPS (10 μg/ml) was added to the mixture and incubated for 4 h at 37°C as described under Materials and Methods. Plasma TNF-α and IL-1β levels were determined by Luminex. TNF-α data are expressed as the mean ± S.E.M. from four separate experiments performed in duplicate, and IL-1β data are expressed as mean ± S.D. from two separate experiments performed in duplicate. IC50 values from these experiments are shown in Table 3.
In addition to a critical role in inflammatory cytokine production, PH-797804 also regulates COX-2 induction and PGE2 production. RASF stimulated with IL-1β produced large amounts of PGE2, with maximal production occurring after 20 h. Complete concentration-dependent inhibition of PGE2 production was observed with RASF treated with PH-797804 (IC50 = 1.5 nM) (Table 3; Fig. 3B). Cell viability was unaffected over the time course studied (data not shown). It is interesting to note that PH-797804 did not affect recombinant COX-2 or COX-1 activity at the concentrations that block PGE2 while inhibiting IL-1β up-regulation of COX-2 mRNA (data not shown), suggesting a unique mechanism for prostaglandin blockade by p38 kinase inhibitors.
The cellular potency of PH-797804 correlates well with its inhibition of recombinant enzyme activity, consistent with a p38 kinase mechanism of action. The U937 cellular activity of PH-797804 was confirmed and extended using LPS-stimulated human monocytes and human whole blood. In monocytes, LPS-stimulated production of both TNF-α and IL-1β was inhibited by PH-797804 in a concentration-dependent manner, with an IC50 value 3.4 nM for each cytokine (Table 3; Fig. 3C), whereas in whole blood, a cellular system that mimics the physiological in vivo milieu, TNF-α and IL-1β production was inhibited, with an IC50 value of 85 and 37.5 nM, respectively (Table 3; Fig. 3D). The lower potency of PH-797804 in human whole blood compared with monocytes and U937 cells is consistent with the plasma protein binding characteristics of the inhibitor (human plasma protein binding, 96.7%) and the hypothesis that only unbound compound is available to penetrate cells. The IC50 values for PH-797804 inhibition of LPS-induced TNF-α and IL-1β production based on the free fraction of the compound in human whole blood were 2.8 and 1.2 nM, respectively, which corresponds with values from both U937 cells and human monocytes.
MAP kinase pathways are also implicated in bone and cartilage destruction in RA by modulating the production and signaling functions of cytokines such as TNF-α, IL-1β, and receptor activator of nuclear factor-κB ligand (RANKL). It is now known that TNF-α and IL-1β function together with RANKL, leading to the propagation of inflammation and bone erosion (Mbalaviele et al., 2006). Using primary rat bone marrow cells, osteoclast formation was induced after 3 days of coculture with RANKL and M-CSF (Fig. 4). PH-797804 inhibited this RANKL- and M-CSF-induced osteoclast formation in a concentration-dependent manner, with IC50 = 3 nM (Table 3; Fig. 4). Because RANKL and M-CSF are required for osteoclast differentiation in vivo, these data suggest that PH-797804 has the potential to inhibit bone destruction associated with inflammatory arthritis.

Morphological view and quantitative analysis of PH-797804 effects on osteoclast formation. Rat bone marrow cells were cultured without (top left) or with RANKL (receptor activator of nuclear factor-κB ligand) (100 ng/ml) and M-CSF (10 ng/ml) (top right) for 3 days in the presence of 0.1 μM PH-797804. At the end of the culture period, cells were stained for TRAP activity. Microphotographs of representative cultures were taken under light microscope at the same magnification. The formation of osteoclasts TRAP+ multinucleated cells with three or more nuclei (arrows) that was induced by RANKL and M-CSF (middle) was blocked by the addition of PH-797804. Rat bone marrow cells were cultured in the presence or absence of RANKL (100 ng/ml) and M-CSF (10 ng/ml). Various concentrations of PH-797804 (0.0001–1 μM) were added to the cultures and the end of the culture period (3 days), cells were stained for TRAP activity, and the numbers of osteoclasts (TRAP+-multinucleated cells with three or more nuclei) were counted. PH-797804 inhibited osteoclast formation in a dose-dependent manner. Results shown are the mean ± S.E.M. of three independent experiments.
PH-797804 Inhibits Acute Inflammatory Responses Induced by Intravenously Administered Endotoxin.
Biochemical models were established to evaluate the oral efficacy and potency of PH-797804 as a blocker of cytokine production in vivo. These models included LPS-induced TNF-α production in rats and cynomolgus monkeys. Intravenous administration of LPS into Lewis rats produced a rapid and transient elevation of TNF-α levels in plasma that peaked 1 to 2 h after LPS injection (data not shown). The extent of inhibition of TNF-α production by PH-797804 was determined by quantifying plasma TNF-α levels of treated and untreated animals. Treatment of rats with PH-797804 resulted in dose-dependent inhibition of LPS-induced TNF-α production (Fig. 5). Plasma levels of PH-797804 were determined 5.5 h after compound administration in parallel with TNF-α concentration determination, and efficacy corresponding to compound concentrations at this time point was determined. Nonlinear, four-parameter analysis of the dose-response and concentration-response data resulted in an ED50 value of 0.07 mg/kg and an EC50 value of 8.6 ng/ml for PH-797804. ED80 and EC80 values were calculated as 0.142 mg/kg and 26.7 ng/ml, respectively (Table 4; Fig. 5).

Effect of PH-797804 on LPS-induced TNF-α production in rats and cynomolgus monkeys as a function of dose (A) and plasma-free fraction (B). Rat: adult male Lewis rats (∼225–250 g) were orally dosed with PH-797804 or vehicle (0.5% methylcellulose and 0.025% Tween 20) 4 h before an intravenous administration of LPS (1 mg/kg). Blood was collected 90 min after LPS challenge. Serum TNF-α levels were quantified by ELISA. Compound blood levels were quantified by liquid chromatography-mass spectrometry. The ED50 value was calculated to be 0.07 mg/kg (A), with an EC50 value of 8.6 ng/ml (18.0 nM). Adjusting for free fraction gives a final EC50 value of 0.4 nM (B). Data are from three separate experiments with five animals per group and are expressed as the mean ± S.E.M.. Monkey: cynomolgus monkeys (n = 3) were dosed intragastrically with PH-797804 or vehicle. Two hours later (time 0), LPS (10 μg/kg) was administered by intravenous injection. Blood samples were taken at 0.75 to 1 h after LPS administration. TNF-α concentrations in the plasma were determined using ELISA. Values are the mean of the percentage of control of peak TNF-α in plasma. Plasma concentrations of PH-797804 were determined by liquid chromatography-mass spectrometry. The ED50 value was calculated to be 0.095 mg/kg (A), with an EC50 value of 7 ng/ml (15.0 nM). Adjusting for free fraction gives a final EC50 value of 0.97 nM (B). Data are expressed as mean values ± S.E.M.
In vivo efficacy of PH-797804 in acute and chronic animal models: LPS-induced TNF-α production in rats and cynomolgus monkeys and rat SCW-induced arthritis
PH-797804 was also orally effective in an analogous model of LPS-induced TNF-α production in cynomolgus monkeys when administered 2 h before LPS challenge (Table 4; Fig. 5). Dose-response analysis resulted in ED50 and ED80 values of 0.095 and 0.349 mg/kg, respectively. The peak exposure of PH-797804 (Cmax) required to produce 50% inhibition of LPS-induced TNF-α production was 7 ng/ml. The dose- and concentration-response analyses demonstrate comparable efficacy and potency for PH-797804 in both the rat and cynomolgus monkey studies (Fig. 5).
PH-797804 Suppresses Chronic Inflammation in Rat Streptococcal Cell Wall-Induced Arthritis and in Mouse Collagen-Induced Arthritis Models.
The ability of PH-797804 to suppress chronic inflammation was demonstrated in both an inflammatory arthritis model in rats induced by SCW extract and in the widely used mouse collagen-induced arthritis model. The SCW model is characterized by an acute phase (hemorrhage and fibrin deposition in the synovial space, accumulation of activated macrophages in the soft tissue, and mild osteolysis) from day 1 to day 5, followed by a more severe and chronic phase (intense cell infiltration, joint inflammation, and bone destruction) that occurs from day 10 to day 21. A role for TNF-α and IL-1β in the chronic phase has been demonstrated with neutralizing TNF-α and IL-1 antibodies (Kuiper et al., 1998).
PH-797804 was highly effective in attenuating SCW-induced inflammation (Table 4; Fig. 6A). PH-797804 treatment resulted in dose-dependent inhibition of paw swelling when administered daily from day 10 to day 21 (a time when untreated control rats exhibited profound joint inflammation), with a maximal efficacy of >95% and an ED50 = 0.186 mg/kg/ EC50 (Cmax) = 203 ng/ml/ EC50 (Cmin) = 32 ng/ml. Doses of 0.5 to 5 mg/kg PH-797804 produced almost complete protection of the joint against inflammation-related bone density loss, whereas doses of 0.05 and 0.15 mg/kg were partially efficacious (Fig. 6B).
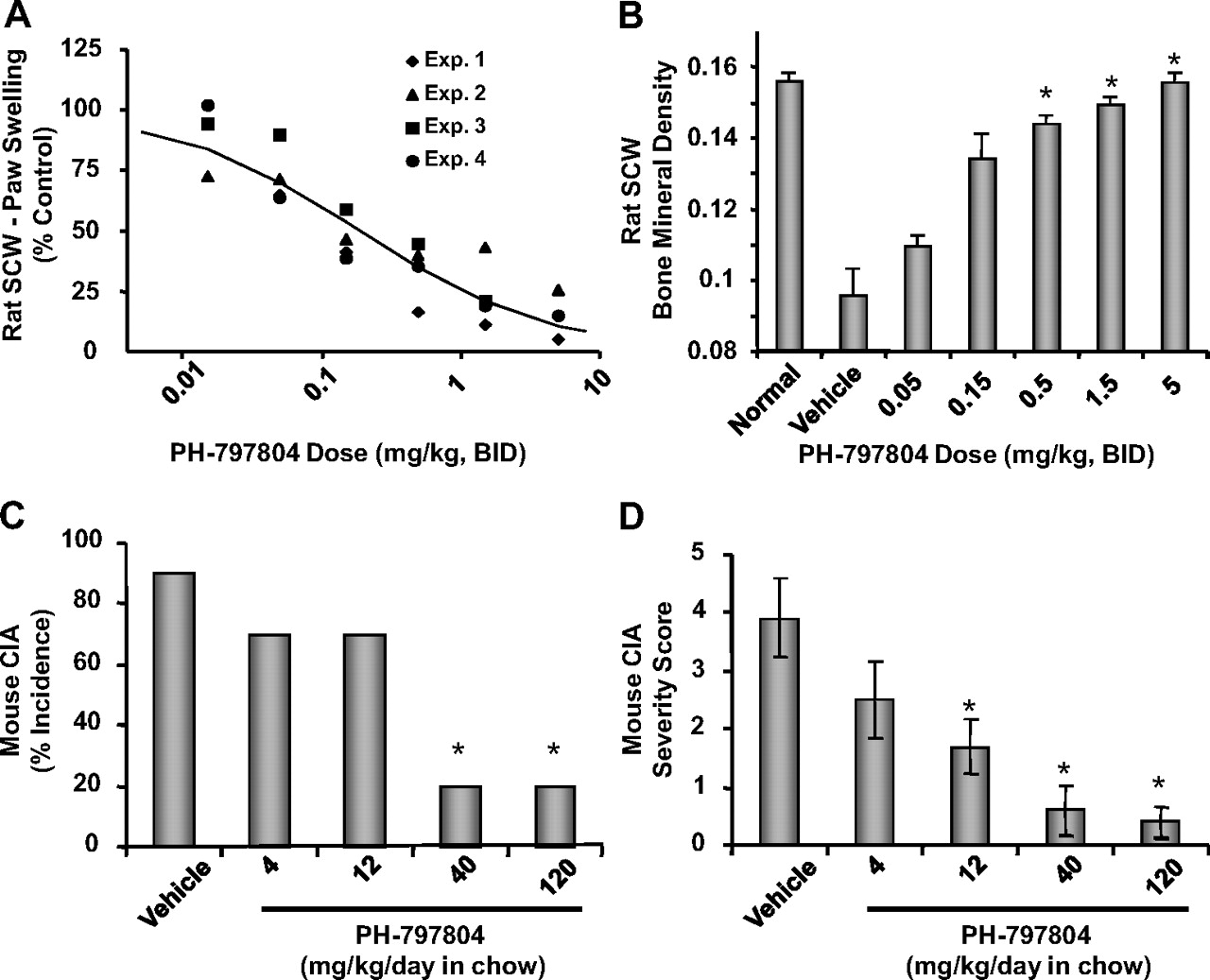
PH-797804 dose-dependently reduces paw inflammation (A) and bone mineral density loss (grams per square centimeter) (B) in rat SCW-induced arthritis and incidence (C) and severity (D) in mouse collagen-induced arthritis. A, rat SCW-induced paw swelling. Administration of PH-797804 from day 10 to day 21 resulted in a dose-dependent decrease in paw swelling. Inhibition of edema was determined by a four-parameter logistical model. ED50 and ED80 values were calculated to be 0.186 and 1.610 mg/kg, respectively. Data are expressed as the mean value obtained from four to eight animals per group, with each of four experiments plotted separately. B, rat SCW-induced bone mineral density loss. Administration of PH-797804 from day 10 to day 21 resulted in a dose-dependent decrease in bone loss measured on day 21. Data are expressed as mean ± S.E.M. from a single experiment with five rats per group. C, disease incidence (mouse CIA). Data are expressed as percentage of animals that demonstrated paw swelling at the termination of the experiment (a severity score of 1 or greater on day 56). D, disease severity (mouse CIA). Animals were evaluated several times per week for signs of arthritis. Scoring of severity was carried out using a score of 1 to 3 for each paw as described under Materials and Methods (maximal score of 12/mouse). Data are the cumulative score determined at the end of the study. Any animal demonstrating a score of 1 or more was considered arthritic. Dose groups consisted of 10 animals per group. *, p < 0.05 (by ANOVA), statistically different from control as described under Materials and Methods.
The mouse CIA model is characterized by the appearance of proliferative synovitis as assessed by paw swelling and redness approximately 3 weeks after immunization with chick type II collagen in Complete Freund's adjuvant. Histological analysis shows infiltration of polymorphonuclear and mononuclear cells into the synovium, cartilage degradation, pannus formation, and bone destruction (Williams, 2007). Treatment of mice from day 21 to day 56 with vehicle demonstrated a 90% incidence of arthritis observed on day 56 (Fig. 6C). Treatment of the animals with PH-797804 at 40 and 120 mg/kg/day in chow resulted in a significant decrease in incidence, with only 20% exhibiting any clinical signs of arthritis (Fig. 6C). This level of inhibition was similar to studies using anti-TNF-α antibody therapy using a similar protocol (Graneto et al., 2007). Lower doses, 4 and 12 mg/kg/day, were less effective in affecting incidence. A dose-dependent decrease in the severity of arthritis was observed at the end of the study, with the 12, 40, and 120 mg/kg/day doses. All three doses were significantly different from vehicle (p < 0.05, ANOVA) (Fig. 6D).
A compilation of enzymatic, cellular, and in vivo data generated on PH-797804 from binding studies to rat SCW-induced arthritis, adjusting for free fraction when appropriate, is shown in Fig. 7. The biochemical efficiency, as defined as the relationship between a functional response (IC50) and the binding affinity of the drug for the target (Ki) (Swinney, 2006), shows a nearly 1:1 correlation across all assays and models. This translatability from enzyme to cell to animal, coupled with human pharmacokinetic projections, resulted in a high level of confidence in the dosage determination of PH-797804 for human clinical studies.

Correlation of functional responses to target binding affinity for PH-797804. A, data for p38α kinase binding, U937 TNF-α production, and human monocyte TNF-α production are each expressed as the mean value of three independent experiments ± S.E.M. Data for U937 p38α kinase activity is as described in Fig 3. B, data for human monocyte IL-1β production and osteoclast formation are each expressed as the mean value of three independent experiments ± S.E.M. Data for RASF PGE2 production are expressed as the mean of six independent experiments ± S.E.M. C, data for human whole blood TNF-α production and human whole blood IL-1β production are the mean/average of four and two independent experiments ± S.E.M./S.D. respectively. Data for rat and cynomolgus TNF-α production and rat SCW paw swelling are as described in Fig 5.
PH-797804 Suppresses LPS-Induced Cytokine Release in Healthy Human Subjects.
A double-blind, placebo- controlled, single-dose study was done to assess the dose and concentration response of PH-797804 on LPS-induced cytokine release in healthy human subjects. In this study, the endotoxin challenge is administered as an intravenous bolus at a final dose of 2 ng/kg. The endotoxin produces a transient endotoxemia evoking mild “flu-like” symptoms that resolve within a few hours. Endotoxin-induced cytokines can be monitored as a measure of the inflammatory response. The objectives of the endotoxemia study were to describe the response of five single-dose levels of PH-797804 (1, 2, 4, 13, and 30 mg) versus placebo on TNF-α and IL-6 blood levels and p38 activity levels.
TNF-α concentrations were measured before study drug dosing on day 1; before LPS administration on day 2; and at 1, 1.25, 1.5, 1.75, 2, 2.25, 2.5, 3, and 5 h after LPS administration. The highest mean TNF-α concentration occurred at either 1.25 or 1.5 h after LPS administration for all groups, with the mean maximal TNF-α concentration for each treatment group being 327.9 pg/ml (placebo), 208.8 pg/ml (1 mg), 188.7 pg/ml (2 mg), 105.8 pg/ml (4 mg), 62.9 pg/ml (13 mg), and 44.5 pg/ml (30 mg). The highest concentrations of TNF-α occurred in the placebo treatment group, consistent with this established human experimental endotoxemia model, whereas PH-797804 induced a dose-dependent inhibition of TNF-α response relationship (Fig. 8A). The data were well described by an inhibitory effect Emax model [E = Emax × (1 − C/(C + EC50))], resulting in EC50 = 2.9 ± 1.2 (0.5–5.3) ng/ml and Emax = 311 ± 35 (240–382) pg/ml [estimate ± S.E. (95% CI)]. The observed data (closed circles) and the predicted concentration-response relationship (solid line) are shown in Fig. 8A. The EC50 concentration along with one approximated S.E. are shown as vertical solid and dashed lines, respectively. As expected in this model, there was a moderate amount of variability (uncertainty over parameter estimate) as reflected by the 40% coefficient of variation on the EC50 parameter estimate.

PH-797804 inhibits LPS-induced TNF-α, IL-6, and MK-2 activity in a dose- and concentration-dependent manner in a human endotoxin challenge model. A, maximal TNF-α concentration versus plasma PH-797804 concentration: individual subject data (closed circles) and predicted response profile (solid line). TNF-α concentrations were maximal levels at each dose group at a time point between 1 h and 3 h after LPS administration. B, cytokine and MK-2 activity inhibition profiles as a function of plasma concentration. Cytokine values are maximal concentrations ± S.E.M.
The affects of PH-797804 administration on LPS-induced IL-6 production and MK-2 activity were assessed as another indication of cytokine modulation and as target modulation, respectively. IL-6 concentrations were measured at pre-LPS administration and 1, 1.5, 2, 2.5, 3, and 5 h after LPS administration. The mean maximal IL-6 concentrations were 620.3 pg/ml (placebo), 370.3 pg/ml (1 mg), 392.2 pg/ml (2 mg), 273.5 pg/ml (4 mg), 289.5 pg/ml (13 mg), and 123.7 pg/ml (30 mg), with the largest mean concentrations occurring at 2 h (placebo and 1, 2, and 30 mg) and 2.5 h (4 and 13 mg) after LPS administration. A concentration response with PH-797804 was observed (Fig. 8B). MK-2 activity, a biomarker for p38 kinase activity, also correlated inversely with PH-797804 dose and showed maximal inhibition at the highest doses and compound blood levels (Fig. 8B). Both IL-6 and MK-2 activity-response relationships correlated well with that of TNF-α, suggesting that administration of PH-797804 24 h before LPS challenge resulted in lower concentrations (relative to placebo) of TNF-α, IL-6, and MK-2 activity in a dose- and concentration-dependent manner.
Discussion
The novel N-phenylpyridinone PH-797804 is a highly selective, readily reversible, ATP-competitive inhibitor of human p38α kinase. Binding potency is primarily driven through two key interactions: 1) engagement of the active site adjacent hydrophobic pocket by the 2,4-difluorophenyl moiety and 2) the formation of a bidentate H-bond between the PH-797804 pyridinone and the M109-G110 peptide backbone in the kinase crossover region (Xing et al., 2009). The relative uniqueness across the human kinome of the large hydrophobic pocket gatekeeper residue (T106) in p38α kinase coupled with the induced peptide backbone flip at G110 required for bidentate H-bond formation are key structural features driving kinase selectivity for PH-797804 (Xing et al., 2009). The p38α kinase isoform-restricted expression in monocytes, U937 cells, and RASF coupled with the high selectivity observed with PH-797804 suggests that the biological activity observed with this compound is driven through modulation of p38α in these cells.
PH-797804 exhibits rapid binding kinetics, consistent with a binding mode that does not engage the allosteric site formed upon the movement of the conserved Asp168-Phe169-Gly170 (DFG) to the “out” conformation. Inhibitors that engage this DFG pocket in p38α exhibit significantly slower association and dissociation binding kinetics (Pargellis et al., 2002) relative to the classical ATP competitive inhibitors. Interactions with this DFG-out pocket in p38α, although generally enhancing compound affinity, may result in decreased selectivity for kinase inhibitors (Angell et al., 2008). Interpretation of p38 kinase-driven biology by using highly selective inhibitors such as PH-797804 is more conclusive than studies using earlier generation p38 inhibitors in which significant kinase crossover was observed (Karaman et al., 2008). Furthermore, the selectivity observed for PH-797804 coupled with a human pharmacokinetic profile exhibiting a low peak/trough ratio (<1.8) resulted in a superior safety profile both preclinically and clinically.
The consistent cellular potency of PH-797804, PH-797804 exhibited by IC50 values in the low nanomolar range across cell types, inflammatory mediator production, and p38 kinase activity measurements is made more interesting by the correlation with the inhibitory potency for the purified enzyme in biochemical test systems. There is essentially a 1:1 correlation between biochemical and cellular potency for PH-797804 across several markers of inflammation (Fig. 7). This high level of biochemical efficiency (Swinney, 2006) is achieved despite the high physiological levels of cellular ATP and the observation that PH-797804 is an ATP-competitive inhibitor. This apparent dichotomy can be rationalized through examination of the relative affinities of ATP and PH-797804 for activated and unactivated p38 kinase. It has been demonstrated previously that ATP binds to activated p38 kinase with much higher affinity than unactivated kinase (Frantz et al., 1998), whereas PH-797804 binds with comparable affinity to both forms of the enzyme. The binding of PH-797804 to the unactivated enzyme in the absence of ATP competition decreases the activation rate of p38 kinase, resulting in the observed biochemical efficiency near unity. A survey of drugs receiving regulatory approval during the current decade indicates that a significant majority exhibit binding mechanisms predicted to achieve high biochemical efficiency, thereby translating to lower dose levels needed to achieve efficacy and resulting in a larger therapeutic index (Swinney, 2006). The potency, selectivity and biochemical efficiency demonstrated by PH-797804 supports this compound as a viable drug candidate for the treatment of inflammatory diseases.
Three of the key hallmarks of RA are pain, joint destruction, and inflammation. We developed cellular and in vivo models to examine the impact of PH-797804 on these key disease features. First, PGE2 is a mediator of inflammatory pain in the arthritic joint. PH-797804 effectively antagonized IL-1β-induced PGE2 production from RA joint synoviocytes, consistent with a potential analgesic role in nociceptive pain. Brief reports of two other p38 kinase inhibitors SCIO-469 (Tong et al., 2004) and Arry-797 (Remmers et al., 2008) described analgesic activity in human clinical studies of dental pain, further supporting the potential utility of p38 kinase inhibitors in this clinical indication.
The second key disease feature investigated is joint destruction. Destruction of bone and cartilage is a hallmark of progressive RA and result in debilitation and morbidity. In addition to attenuating paw edema, significant protection of joint bone mineral density was observed with PH-797804 in the SCW-induced arthritis model, consistent with studies on earlier p38 kinase inhibitors (Mbalaviele et al., 2006). Studies using rat bone marrow osteoclasts suggested that the mechanism by which PH-797804 exerts its bone protective effect may be through inhibition of osteoclast differentiation, the cell type primarily responsible for bone resorption (Roodman, 1999). Treatment of rat bone marrow cells with PH-797804 resulted in a concentration-dependent inhibition of osteoclast formation induced by the inflammatory cytokines RANKL and M-CSF, with a potency comparable with its ability to inhibit TNF-α production and p38 kinase activity in other cell-based assays.
Third, from an inflammation standpoint, the significant efficacy of anticytokine biologic therapy in RA has demonstrated a role for cytokines such as TNF-α, IL-1, and IL-6 as central mediators of the inflammation associated with the disease. Numerous studies have demonstrated that p38 kinase is a key enzyme in the intracellular networks that transmit an inflammatory signal from the cell surface to trigger cytokine production. PH-797804-induced blockade of TNF-α and IL-1β production from human monocytes and whole blood support a role for p38 kinase in the biosynthesis and release of these cytokines. The inhibitory potency of PH-797804 is consistent across cell models and cytokine measurements, with IC50 values in the 3 to 6 nM range (taking into account nonprotein bound drug in human whole blood as the active fraction). Furthermore, the corresponding inhibition of p38 kinase activity in these cells is consistent with a mechanism of action associated with specific antagonism of this kinase.
PH-797804 efficacy in modulating cytokine production in endotoxin challenge models in the rat and cynomolgus monkey was comparable across these two species and corresponded to efficacy observed in human cellular models. In addition, concentration-response analysis of these data demonstrated compound potency comparable to human cell activity of PH-797804. EC50 values for rat and cynomolgus monkey inhibition of endotoxin induced TNF-α production were 0.34 and 0.92 nM, respectively, based upon drug-free fraction (PH-797804 plasma protein binding was 98.1 and 93.7% in these two species, respectively). The similar potency observed across cellular and acute in vivo models provided confidence in the human efficacious dose projection based upon concentrations of PH-797804 needed modulate p38 kinase activity and TNF-α production in these models.
Further evidence supporting anti-inflammatory properties of PH-797804 was demonstration of efficacy in blocking paw edema in two rodent models of RA, rat SCW and mouse CIA, after oral dosing. It is interesting that in rat SCW, effect on paw edema directly correlated with Cmin values for PH-797804, defined as blood concentration of drug 12 h after administration of compound (Fig. 8C; in this model, PH-797804 is dosed twice a day for 10 days). These observations suggest two pharmacodynamic properties of PH-797804. 1) Persistent inhibition of p38 kinase activity is required to achieve anti-inflammatory efficacy in the SCW arthritis model, and 2) the potency of PH-797804 for achieving anti-inflammatory efficacy in a subchronic model of RA is comparable to that required in acute in vivo and cellular models.
The potency of PH-797804 in nonclinical efficacy studies together with human pharmacokinetic projections was used to determine the doses of PH-797804 for the human endotoxemia study. Doses bracketed the range projected to generate blood concentrations of drug that spanned 0 to 95% p38 kinase inhibition and consequent cytokine modulation. As a result of slow drug absorption and long half-life, endotoxin challenge was administered 24 h after PH-797804 dosing, a time in which maximal drug levels were observed. A complete dose and concentration response was achieved, resulting in a maximal 80 to 90% inhibition of TNF-α and IL-6. As expected, inhibition of p38 kinase activity was complete at the higher doses and correlated with cytokine regulation. The effect of p38 kinase inhibition on cytokine production in human endotoxemia has been reported previously (Branger et al., 2002; Faas et al., 2002); however, experiments described here are the first that demonstrate quantitative preclinical-to-clinical pharmacokinetic and pharmacodynamic translation of inhibitor effect on the target (p38 kinase) and key inflammatory mediators. These experiments further demonstrate the utility of cellular and/or acute inflammation models as predictors of human activity.
The potential anti-inflammatory therapeutic utility of p38 kinase inhibition remains an open question. Although the first wave of p38 kinase inhibitors that were evaluated clinically demonstrated development-limiting toxicities, reports of newer generation, more selective compounds suggest less of an issue with adverse events. It is interesting that recent reports suggest that the anti-inflammatory efficacy of p38 kinase inhibition in RA was not robust or sustained (Genovese et al., 2008; Cohen et al., 2009; Damjanov et al., 2009). Lack of information on pharmacokinetic and target coverage information on these compounds precludes a mechanistic understanding of this observation. Overall, the potency, selectivity, biochemical efficiency, in vivo efficacy, and pharmacokinetic properties of PH-797804 make it a strong candidate to definitively evaluate the role of p38 kinase in human inflammatory disease. Toward that end, PH-797804 is under evaluation in phase 2 clinical studies of RA and chronic obstructive pulmonary disease.
Acknowledgments
We thank Gail Jungbluth and Marie-Pierre Hellio Le Graverand-Gastineau for contributions to the clinical studies and Robert Chott for technical assistance.
Footnotes
-
↵1Current affiliation: Millipore Corporation, St. Charles, Missouri.
-
↵2Current affiliation: Monsanto Company, St. Louis, Missouri.
-
This study was sponsored by Pfizer Inc.
-
Portions of this work were presented at the following conference: Monahan J, Hope H, Schindler J, Jungbluth G, Burnette B, Guzova J, Hirsch H, Saabye M, Compton R, Zhang J, et al. (2009) Anti-inflammatory properties of a novel N-phenyl pyridinone inhibitor of p38 MAP kinase: preclinical to clinical translation. Annual European Congress of Rheumatology; 2009 Jun 10–13; Copenhagen, Denmark, EULAR, Zurich, Switzerland.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.109.158329
-
ABBREVIATIONS:
- RA
- rheumatoid arthritis
- TNF
- tumor necrosis factor
- IL
- interleukin
- COX
- cyclooxygenase
- MAP
- mitogen-activated protein
- ERK
- extracellular signal-regulated kinase
- JNK
- c-Jun NH2-terminal kinase
- PH-797804
- benzamide, 3-[3-bromo-4-[(2,4-difluorophenyl)methoxy]-6-methyl-2-oxo-1(2H)-pyridinyl]-N,4-dimethyl-, (−)- (9CI)
- SCW
- streptococcal cell wall
- LPS
- lipopolysaccharide
- MKK
- mitogen-activated protein kinase kinase
- EGFRP
- epidermal growth factor receptor peptide
- GST
- glutathione transferase
- RASF
- rheumatoid arthritis synovial fibroblast(s)
- MK-2
- mitogen-activated protein kinase-activated protein kinase 2
- PG
- prostaglandin
- ELISA
- enzyme-linked immunosorbent assay
- M-CSF
- macrophage–colony-stimulating factor
- TRAP
- tartrate-resistant acid phosphatase
- TRAP+
- tartrate-resistant acid phosphatase-positive
- HSP
- heat shock protein
- Arry-797
- Arry-371797 (N-substituted-5-(2,4-difluorophenoxy)-1-isobutyl-1H-indazole-6-carboxamide)
- CIA
- collagen-induced arthritis
- CII
- chick type II collagen
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- SB203580
- 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole
- RANKL
- receptor activator for nuclear factor-κB ligand
- DFG
- Asp168-Phe169-Gly170.
- Received July 17, 2009.
- Accepted August 28, 2009.
- © 2009 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}