





Abstract
Respiratory syncytial virus (RSV) is known to cause acute lower respiratory tract infections (ARI) in young children and is involved in exacerbation of chronic obstructive pulmonary disease (COPD) in adults. The role of RSV in stable COPD and the viral load in different respiratory diseases has not been investigated to date.
The present authors established and evaluated a quantitative TaqMan® real-time polymerase chain reaction assay specific for RSV subgroup A. Absolute quantification for the determination of viral load input was achieved using a control plasmid. The assay allowed for a quantification over a >6-log range and a detection limit of <10 RSV copies per reaction mixture.
The assay was 30 times more sensitive than conventional nested polymerase chain reaction assays. Interassay sd was 1.3 and coefficient of variation 4.7% on average. Clinical specimens from infants with ARI (n=62) and elderly adults with COPD (n=125) were compared for viral loads. A total of 47% RSV-positive samples were found in the ARI study group and 28% in the COPD study group. The viral load of both study groups was found to differ significantly. In the ARI study group the viral load was increased almost 2000-fold, suggesting acute infection in this group and former or latent infection in the COPD group.
Respiratory syncytial virus-A specific TaqMan® real-time polymerase chain reaction assay is a sensitive and rapid method for the determination of viral load in clinical samples. It enables differential statements concerning the involvement of respiratory syncytial virus in acute lower respiratory tract infections and chronic obstructive pulmonary disease to be achieved.
- bronchiolitis
- chronic obstructive pulmonary disease
- pulmonary diseases
- real-time polymerase chain reaction
- respiratory syncytial virus
- viral load
This study was supported by the Bundesministerium für Bildung und Forschung (BMBF) grant 01GC-9802/8 and 01GC-0101.
Human respiratory syncytial virus (RSV) is one of the most important and frequent viruses for respiratory tract infections. Worldwide RSV causes severe lower tract infections like bronchiolitis or pneumonia in infants and young children 1 and is a common cause for hospitalisation. An association between primary RSV infection and chronic abnormalities ofpulmonary function, especially childhood asthma, can be suggested as a result of a long-term prospective study in children after RSV infection 2. RSV is also common in adults, but usually causes mild upper respiratory tract disease. However in certain adult populations, for instance elderly adults and/or adults with chronic obstructive pulmonary disease (COPD), RSV can cause serious lower respiratory tract infections 3.
RSV is an enveloped ribonucleic acid virus of the genus Pneumovirus within the family Paramyxoviridae. Two antigenically distinct subgroups, group A and B, are known 5. Epidemiologic studies have shown that there are three types of RSV epidemics, those in which group A or group B viruses were dominant and those in which both groups circulate concurrently 6. In children subgroup A strains were detected at least three times as often as subgroup B in most years 7. Moreover it has been shown that the course of disease for infections with RSV strain A is usually more severe 8.
A diagnostic method used increasingly for the detection of viral pathogens, as causes for infections, is the polymerase chain reaction (PCR). Compared with the traditionally used standard laboratory methods, viral culture or enzyme-linked immunosorbent assays, PCR not only achieved higher specificity and sensitivity 10–12, but it also facilitated practical performance, particularly in screening large numbers of samples 11. There are many different PCR techniques presently in use. Until recently PCR was used mainly in a qualitative fashion by visualising the amount of amplified target molecules at the end of the reaction (conventional PCR). Sensitivity of such assays can be increased with a nested PCR (nePCR), where samples with very few starting viral genomes are analysed, but this requires a second time-consuming PCR run and increases the risk of cross-contamination. Quantitative real-time PCR (qPCR) based on the 5′-3′ exonuclease activity of Thermus acquaticus (Taq) polymerase on the other hand has shown increased sensitivity and specificity 14–16. Due to the measurement of theamplification product in the exponential phase of the reaction, differences in the amount of starting viral molecules can be detected 15 and the initial concentration can be measured in a definite volume (viral load). The additional information of viral load can be a useful diagnostic tool to predict virus-associated diseases, assess disease status, identify different states of viral infection or monitor the efficacy of antiviral therapy 18–23. The results of qPCR experiments, which usually need <2 h, can be analysed directly without any post-PCR steps.
The aim of the current study from members of the Clinical Research Group “Viral infections in acute and chronic respiratory disease of children and adults” was to evaluate a sensitive and rapid RSV-A specific qPCR assay for the determination of viral load in different respiratory diseases. Specimens of hospitalised children with acute respiratory tract infection (ARI) and of adults with COPD were analysed for the presence and quantity of RSV-A. The second aim of the present study was to determine whether RSV-A could be found in COPD patients with and without signs of acute exacerbation by using a highly sensitive method.
Materials and methods
Virus stock and culture
RSV subgroup A Long strain (ATCC number VR-26) andHEp-2 host cells (ATCC number CCL-23) were kindly provided by the Institute of Medical Microbiology and Virology (Ruhr-University Bochum, Germany). After incubation of 72 h the virus supernatant was titrated, by serial dilution and the 50% tissue culture infective dose (TCID50) was calculated using the Kärber formula. RSV stock was divided into aliquots and stored at −70°C.
Patients and respiratory specimens
Clinical specimens were obtained from two study groups. In the first study group nasopharyngeal secretions were collected between January and June 2001 from 62 consecutive children hospitalised for ARI by suction of both nostrils. All patients aged up to 36 months and presenting with primary (n=54) or nosocomial (n=8) infection of lower airway were included. Diagnoses were as follows: spasmodic croup, bronchitis, wheezing bronchitis, bronchiolitis and pneumonia. Exclusion criteria consisted of existing contraindications of nasal suctioning and immunosuppressive therapy. Patients with primary or secondary immunodeficiency or congenital heart disease were not included. Nine of the 29 RSV-positive patients (31%) and five of the 33 controls (15%) were born prematurely (<35 weeks gestational age). All specimens were obtained by a current method 24 at the Children's Hospital Bochum (Bochum, Germany) and stored at −70°C.
The second group consisted of 125 hospitalised elderly patients (median age 69 yrs, range 43–81 yrs) with COPD. A total of 79 of these elderly patients were hospitalised for acute exacerbation of COPD (AE-COPD) characterised by the following: worsening in dyspnoea, cough and expectoration. The remaining 46 elderly patients had stable COPD and had been hospitalised for other medical reasons. These patients had suffered no exacerbation within the last 30 days prior to their hospitalisation and had undergone no changes in their therapy within the last 14 days (including inhaled and oral medication). Both COPD groups had moderate disease (Global Initiative for Chronic Obstructive Lung Disease: GOLD II) 25. The most frequent comorbidities in the AE-COPD group were as follows: hypertension (45/79, 57%), coronary artery disease (23/79, 29%), hyperlipoproteinaemia (20/79, 25%) and diabetes (18/79, 23%). In the stable COPD patients the most prevalent comorbidities were: hypertension (27/46, 59%), hyperlipoproteinaemia (16/46, 35%), coronary artery disease (10/46, 22%) and diabetes (7/46, 15%), none of these patients were immunodeficient. Nasal lavage fluid and induced sputum were collected between October 1999 and October 2001 at the Dept of Internal Medicine, (University Hospital Bergmannsheil, Bochum, Germany). All specimens were obtained by the method described by Rohde et al. 26. Samples were subjected to centrifugation and both cells and cell-free supernatants were frozen at −70°C.
Ribonucleic acid extraction and reverse transcription
RNA isolation procedure was carried out immediately after thawing using 250 µL of cells (RNeasy Mini Kit; QIAGEN, Hilden, Germany) and 1000 µL of cell-free supernatant (QIAamp DNA Blood Mini Kit; QIAGEN) according to the manufacture's instructions. RSV-infected HEp-2 cells and RSV stock supernatant used as a control were processed in the same way as cell samples and cell-free supernatants. The final volumes of isolated RNA were 150 µL for cell samples and 100 µl for cell-free supernatants. All samples were aliquoted and stored at −70°C. Complementary deoxyribonucleic acid (cDNA) was synthesised with 2.5 µM random hexamers using 10 µl extracted RNA in a final volume of 50 µl by using TaqMan® Reverse Transcription Reagents kit (Applied Biosystems, Foster City, CA, USA). The cDNA samples were stored at −20°C.
Design of primers and probe
Forward and reverse primer and probe sequences were designed with Primer Express software (Primer ExpressTM Version 1.0; Applied Biosystems, USA) with regard to general rules of primer and probe design. The present primer and probe system (table 1⇓) is located within a RSV genome region encoding for the F1 subunit of the fusion protein and the predictive amplicon length is 76 base pairs (bp). A search alignment was done confirming specificity of primers and probe for RSV-A Long strain. The probe contained the fluorescent reporter dye 6-carboxyfluorescein (FAM) at the 5′-end and the fluorescent quencher dye 6-carboxytetramethylrhodamin (TAMRA) at the 3′-end. Primers and probe (Applied Biosystems, Darmstadt, Germany) were aliquoted to avoid lost of stability by freezing and thawing and stored at −20°C.
Sequences of oligonucleotides used as primers and probe in respiratory syncytial virus (RSV)-A specific quantitative real-time polymerase chain reaction assay
Quantitative real-time polymerase chain reaction
PCR reactions were carried out as reverse transcriptase (RT)-qPCR with 5 µL cDNA in a volume of 25 µL with TaqMan® PCR Core Reagent Kit (Applied Biosystems, USA). Primers and probe were used at a concentration of 300 nM and 200 nM respectively. For all PCR amplifications positive and negative controls were included. Positive controls consisted of nucleic acid extracted from RSV supernatant and/or plasmid standard deoxyribonucleic acid (DNA). Negative controls were carried out with water instead of RNA. PCR runs were carried out according to the standard TaqMan® PCR profile. Amplification of target DNA and detection of PCR products were performed with a GeneAmp® 5700 Sequence Detection System (Applied Biosystems, USA). Amplification of the target sequence was detected by an increase of fluorescence above a baseline with no or little change in fluorescence. In order to analyse data, the reporter (FAM) fluorescence was automatically normalised to a passive reference to avoid the measurement of non-PCR-related fluorescence. A threshold was set above the baseline and a threshold cycle value (Ct) was defined as thecycle number at which the fluorescence passes the fixed threshold and a statistically significant increase in fluorescence is first detected.
Nested polymerase chain reaction
The nePCR was carried out using RSV subgroup A specific primer sequences as described by Rohwedder et al. 27 and QIAGEN Taq Polymerase Protocol (QIAGEN). All nePCR reactions were performed as RT-PCR.
Hexaplex® Multiplex reverse transcriptase polymerase chain reaction
All multiplex RT-PCR reactions were carried out at the Institute of Medical Microbiology and Virology, Ruhr-University (Bochum, Germany) using the Hexaplex® Multiplex RT-PCR system (Prodesse, Waukesha, WI, USA).
Enzyme immunoassay
Enzyme immunoassay detection of RSV was made with ABBOTT testpack RSV (Abbott, Wiesbaden, Germany) and carried out at the Children's Hospital of the Ruhr-University (Bochum, Germany).
Exclusion of cross reactivity
Respiratory tract specimens positive for Rhinovirus, Influenza A or Parainfluenza 3, or positive for multiple infections with these viruses and RSV (analysis by nePCR or multiplex PCR) were chosen to study qPCR specificity.
Quantitative real-time polymerase chain reaction product analysis and construction of a standard
The predictive length of the qPCR amplification product was confirmed by a single clear band of 76 bp length on an ethidium bromide-stained 4% NuSieve® agarose gel (FCM BioProducts, Rockland, ME, USA) under ultraviolet illumination. Additionally PCR products were cloned with the TOPO TA Cloning® Kit for Sequencing (Invitrogen, Karlsruhe, Germany) into the vector pCR4®-TOPO and transformed into an appropriate bacterial strain (TOP10). The obtained clones were evaluated by qPCR to confirm the presence of a plasmid containing the RSV insert. Isolation of plasmid DNA was carried out with the QIAprep Spin Miniprep Kit (QIAGEN) and concentration was calculated from optical density measurements at 260 nm in a Beckman Du®-70 spectrophotometer. Plasmid DNA was sequenced with BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, USA) on a ABI PRISMTM 310 Genetic Analyzer (Applied Biosystems, USA).
Absolute quantification
For standardisation of the assay the plasmid DNA (pCR4-T1/6) isolated from positive-prescreened clone T1/6 was used. The concentration was calculated accurately from optical density measurements and copy numbers of plasmid DNA were calculated by multiplying the product of concentration (pM·µg−1) and dilution (µg·µL−1) with the Avogadro constant. Serial 10-fold dilutions of known quantities, ranging from 10 pg·µL−1 to 1×10−4 pg·µL−1 corresponding to 4.8×106–48 RSV-copies·µL−1 were made, aliquoted and stored at −20°C. The results of 22 measurements of each standard concentration were used for the design of a general standard curve for mean Ct values in order to calculate copy numbers in samples of different study groups when means had to be compared (see Results section).
Statistical analysis
The Applied Biosystems ABI PRISMTM Sequence Detection System, was used to monitor the increase of the reporter fluorescence during PCR and the entire process of calculating Ct values, preparing a standard curve and determining the starting copy numbers for unknown samples was performed by the software. Data were presented as amplification plot, showing the fluorescent values plotted versus the cycle number and as standard curve, displaying the threshold cycle versus the logarithm of defined quantities (copy numbers) of the constructed plasmid standard samples. Copy numbers ofunknown samples were automatically inferred from theregression line. Viral load values are declared as copy numbers per PCR reaction mixture and copy numbers per mL, respectively in the case of clinical specimens. To evaluate reproducibility, intra-assay and interassay sds and coefficients of variation (CVs) were calculated for each standard concentration within and between individual PCR runs. To compare results achieved from different study groups the Kolmogorov-Smirnov test for normality was used. Mann-Whitney Rank Sum test was used in case of failed normal distribution. For all tests significance level of 5% was chosen.
Results
Evaluation of respiratory syncytial virus subgroup A specific quantitative real-time polymerase chain reaction
The qPCR amplifications of standard plasmid DNA were performed showing amplification plots corresponding to mean Ct values of 17.66–36.82 (fig. 1⇓). For absolute quantification of viral genomes a standard curve was designed from Ct values plotted versus the log of standard concentrations (fig. 2a⇓). The authors found a highly significant linear relationship between the log of the input target DNA copy numbers and Ct values, thus admitting interpolation of the input RSV concentration (viral load) of samples containing unknown quantities of RSV-RNA. Based on the standard curve the limit of detection of viral RNA was eight RSV copies per reaction mixture (table 2⇓).

Amplification plots of pCR4-T1/6-standard deoxyribonucleic acid. Ten-fold serial dilutions were made with 4.8×106 to 48 respiratory syncytial virus copies per reaction mixture. These representative quantitative TaqMan® real-time polymerase chain reaction (PCR) amplification plots show the target sequence amplification as increase of the fluorescence reporter signal (Rn) versus the cycle number during a PCR run. The threshold cycle values are calculated automatically by determining the point at which the fluorescence exceeds a fixed threshold line (═).

a) A standard curve for pCR4-T1/6-standard deoxyribonucleic acid (DNA) with input concentrations (Log CO) of 10 to 1×10−4 pg per reaction mixture. Unknown respiratory syncytial virus (RSV) genome quantities of clinical samples were determined by the ABI PRISM® 5700 Sequence Detection System software by interpolation from the standard curve. b) Standard curve of six pCR4-T1/6-standard DNA dilutions evaluated as mean value standard curve. Each point represents the mean threshold cycle (Ct) value and was calculated from the results of the 22 polymerase chain reaction amplifications.
Correlation between respiratory syncytial virus (RSV) detection in RSV-ribonucleic acid dilutions determined by quantitative real-time polymerase chain reaction (qPCR) and nested PCR (nePCR)
To determine precision and reproducibility of the present assay the Ct values obtained from 22 replicates of each standard dilution were analysed. The sds ranged from 0.58–1.71 for intra-assay runs and from 0.56–1.95 for interassay runs, with higher values found for lower copy number dilutions (table 3⇓). These results allowed the design of a general standard curve for mean Ct values, in order to calculate copy numbers in samples of different study groups (figure 2b⇑). The higher sd value for the lowest concentration corresponds to sensitivity findings of the assay which was reduced to 77.3% in the range of 48 copies per reaction mixture. Although precision of quantification below this copy number may become slightly inaccurate, the authors' qPCR detected <48 RSV copies per reaction mixture (table 2⇑). Intra-assay and interassay CVs were 4.15% (range 3.1–5.4%) and 4.68% (range 3.2–5.9%) on average and showed no significant difference.
Interassay precision (repeatability) of respiratory syncytial virus (RSV)-A specific quantitative real-time polymerase chain reaction
Respiratory syncytial virus detection by quantitative real-time polymerase chain reaction and nested polymerase chain reaction
The sensitivity of the qPCR primers and probe system was tested with cDNA generated from RSV stock. Tenfold serial dilutions of isolated RSV-RNA corresponding to a TCID50 of 1.2×104 to 1.2×10−9 were used, reverse transcribed and analysed by qPCR. The authors found successful amplification of RSV-cDNA in the first ten dilutions, until TCID50 of1.2×10−5. Ct values spread over the measuring range from Ct 15.21–Ct 39.44, corresponding to computed copy numbers of 1.67×107 copies per reaction mixture to 8.43 copies per reaction mixture.
The results of the authors' assay were compared with those achieved by a well established nePCR for RSV detection described by Rohwedder et al. 27. Positive nePCR amplification results in a 326 bp PCR product that could be detected as asingle band by agarose gel electrophoresis. Both PCR protocols were applied to the same RNA dilutions from RSV stock as described above. The results of both PCR methods were corresponding for the first eight dilutions of RSV-RNA, but nePCR detection stopped at TCID50 of 1.2×10−4, whereas qPCR was more sensitive by two logs (table 2⇑).
Study of a respiratory syncytial virus-infected human cell line
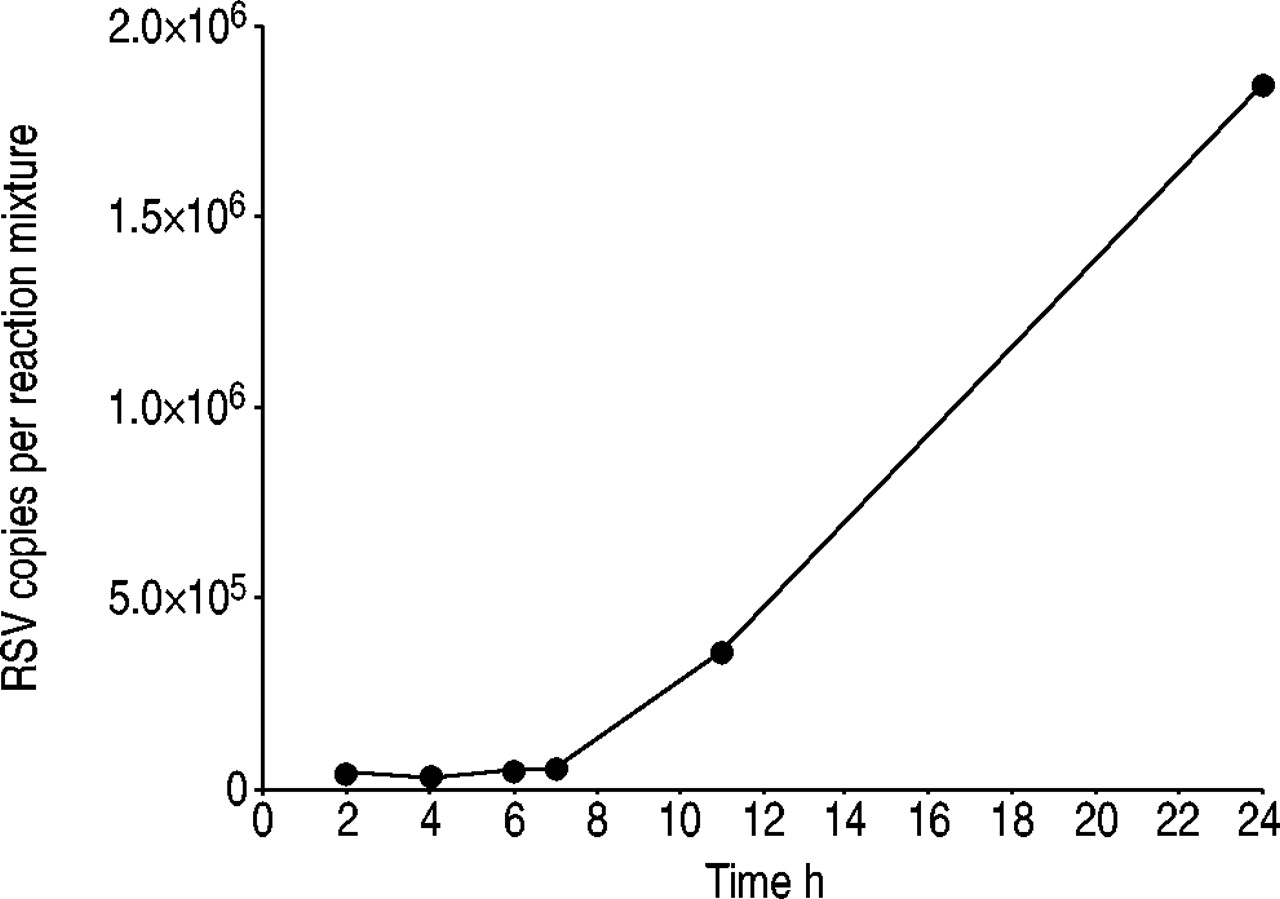
RSV-infected HEp-2 cells were studied in order to examine the ability of the authors' assay to detect RSV-RNA, specifically and sensitively in a complex mixture with human RNA. HEp-2 cells were RSV-infected with different multiplicities of infection (moi 1×10−2–1×10−6) under a constant incubation time of 2.5 h. RNA was isolated, reverse transcribed and the qPCR protocol was applied to all samples. The assay was found to be successful in detecting different amounts of virus nucleic acid in RSV-infected HEp-2 cells, corresponding to different multiplicities of infection (data not shown). Moreover viral replication in HEp-2 cells could be demonstrated. Figure 3⇓ shows the increase of RSV-RNA in infected HEp-2 cells (moi 0.01) at six time points within 24 h of incubation. The authors' qPCR assay indicated that at the end of the incubation time, the viral load of RSV-infected HEp-2 cells had increased 43-fold compared with the value measured after 2 h of incubation.

Increase of respiratory syncytial virus (RSV) genomes after RSV propagation in HEp-2 cells. HEp-2 cells had been infected with RSV (multiplicity of infection 0.01) and incubated for 24 h at 37°C. At six time points incubation had been stopped, ribonucleic acid was isolated from HEp-2 cells, reverse transcribed and quantitative real-time polymerase chain reaction amplification was carried out. The RSV copy number after 2 h of incubation was 4.3×104 per reaction mixture, increasing to a 43-fold number of viral genomes (1.8×106 copies per reaction mixture).
Analysis of the qPCR-amplification product by agarose gel electrophoresis showed a single clear band corresponding to the predictive length. Subsequent sequencing of the PCR product confirmed specificity for RSV-A Long strain.
Cross-reactivity with other human respiratory viruses
In order to further analyse specificity of the assay the current authors' investigated whether other human respiratory viruses (Rhinovirus, Parainfluenza-3 virus and Influenza-A virus) were detected by the qPCR protocol or inhibited RSV detection when multiple virus infections were present. For this purpose patient samples with confirmed non-RSV infection were studied. Neither Rhinovirus-, Parainfluenza-3 virus- or Influenza-A virus-positive specimens yielded any PCR-amplification with RSV-A specific qPCR primers and probe. Moreover no tested specimens with confirmed multiple infection failed to yield a positive PCR result whenever RSV was present.
Study of clinical samples by respiratory syncytial virus-A specific quantitative real-time polymerase chain reaction
The qPCR detection of RSV in clinical samples was studied using nasopharyngeal secretions of infants with ARI (n=62). A total of 29 (46.8%) of these 62 specimens (cell samples) were positive for RSV-A, and 33 samples did not show any amplification in the PCR reaction. Furthermore the authors' qPCR assay provided information on the quantity of RSV nucleic acid in the ARI study group. The viral load found in these infant respiratory samples ranged between 5.4×103–8.5×108 copies·mL−1 (median 1.2×107 copies·mL−1). Additionally the qPCR results of the ARI group were compared with the results of two other diagnostic tests, the Hexaplex® Multiplex RT-PCR and an enzyme-immunoassay test for rapid RSV antigen detection. Both tests detect RSV group A and group B but only the multiplex PCR system can differentiate both strains. To analyse specificity and sensitivity of the qPCR assay the results were compared with those specimens positive or negative for both, the multiplex and the antigen test (n=43), defined as a standard. Specificity and sensitivity of RSV-A specific qPCR were 95% and 91.3%, respectively.
In a second study group of elderly hospitalised COPD patients RNA was extracted from 125 nasal lavages and sputum cell samples. A total of 35 samples (28%) were detected to be RSV-A positive (27.9% in the AE-COPD and 28.3% in the COPD subgroup). There was no significant difference between the viral load of positive nasal lavage samples and the viral load of positive sputum samples. The viral load in this second study group differed significantly from the viral load results obtained from the child study group (fig. 4⇓). The median viral load of the respiratory specimens of the COPD study group was 6.1×103 copies·mL−1 (range 3.2×103–1.5×107 copies·mL−1), reduced 1967-fold compared to the ARI study group. The authors found as few as 15 RSV genomes (median) on average per reaction mixture in respiratory samples of the COPD study group showing that the qPCR is able to detect even very low copy numbers of viral nucleic acid molecules in clinical samples.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Respiratory syncytial virus (RSV) viral loads of clinical samples analysed by quantitative real-time polymerase chain reaction. RSV viral loads are shown for respiratory specimens taken from hospitalised children with acute respiratory tract infections (ARI; n=62) and of elderly chronic obstructive pulmonary disease (COPD) patients (n=125). A total of 29 and 22 specimens were found to be RSV-positive in the ARI and the COPD groups, respectively. The median number of 1.2×107 RSV-genomes·mL−1 was found in the ARI group (range 8.5×108–5.4×103 RSV-genomes·mL−1) significantly higher than the median number of 6.1×103 RSV-genomes·mL−1 found in the COPD group (range 1.5×107 to 3.2×103 RSV-genomes·mL−1). ***: p<0.001.
In addition the authors studied cell-free sample supernatants with the qPCR assay and compared the results with those of cell samples. In the ARI group positive and negative results of cell and cell-free samples corresponded in 100% (n=62/62) whereas in the COPD group correspondence was only 17% (n=82/125).
Discussion
In the present study an RSV-A specific qPCR assay was evaluated and applied to clinical samples. RSV is the main cause of childhood viral ARI 28 and has been detected among other respiratory viruses, mainly influenza A, rhinovirus, coronaviruses and parainfluenza viruses, in exacerbations of COPD 29–32. For older adults it has been shown that the diagnosis of RSV with less sensitive tests can be difficult and are not always reliable, leading to the complementary use of different methods for RSV detection 33. The main advantages of the RSV-A specific qPCR assay are high specificity and sensitivity as well as its exact quantification and reproducibility. The detection of RSV-A was reliable both in respiratory samples of children and in those of elderly patients. Comparison of qPCR results to data obtained by nePCR revealed an almost 30-fold increase in sensitivity, corresponding to results of other studies 35. Moreover minimal assay variation represented by a mean CV of 4% of standard plasmid DNA was found compared with 14% reported for conventional PCR assays 36.
The importance of respiratory viruses in exacerbations ofCOPD has been shown in a number of studies using less sensitive methods 29 and in studies using PCR technology 26. Moreover viral pathogens have been documented in COPD without worsening of respiratory status 26. RSV-A was found in the present study at similar percentages in the COPD study groups with and without exacerbation (27.9 and 28.3%, respectively). The implication of RSV and other viruses in COPD has recently been studied by Seemungal et al. 37 by nePCR, whose results confirm a high number of RSV-positive specimens (23.5%) among stable COPD patients. In a previous study RSV could not be detected in this group using nePCR technology 26. Greenberg et al. 32 found RSV detection rates in documented respiratory tract viral infections of 11.4% for COPD and 9.9% for control patients by using viral serology. Due to the high sensitivity of the qPCR assay, very low viral load values can be detected, probably not included in studies using less sensitive methods. However a comparison of results from diverse studies is onlypartly possible because of differing study design, study population, definition of illness and detection methods with different specificity and sensitivity.
The main goal of the present study was to quantify exactly the amount of viral particles in clinical samples and to compare different respiratory diseases by analysing viral loads. In recent years viral load assays have been established and successfully applied for several infectious diseases 18–23 even as a criterion for distinguishing between acute and latent infection 39. The present study's results show that the RSV-A specific qPCR is able to detect and to quantify a wide range of different viral loads in respiratory tract specimens. Because of the genetic heterogeneity of both RSV subgroups detection of RSV-B by a RSV-A specific qPCR is excluded. This limits the method to detection of RSV-A only, but since RSV-A is more prevalent 7–9 it does not limit the interpretation of the data presented here. Moreover the assay outlined in this study allows for a differentiated statement on the implication of RSV in different diseases. The authors found a significant difference between the mean of RSV-copies detected in children with ARI compared to COPD patients. In the child-ARI group the authors found very high viral loads. These children suffered from a disease which was acute in its onset and required hospitalisation. At this very special time point the samples were collected and analysed. Combining these findings and considering the additional diagnostic methods used the authors have no doubt that these children suffered from acute infection with RSV-A. In the COPD group, however the authors found two different clinical presentations. One suffered from an acute exacerbation whereas the other showed a stable disease. Although one subgroup was exacerbated and needed hospitalisation the authors found the same low viral loads in both groups without significant differences. As observed in the child study group, it would be expected that high viral loads are associated with acute infection. Therefore the authors hypothesised that low viral loads indicated a latent infection or a small amount of residual RSV genome from a previous infection. Given the low viral loads found in AE-COPD group the authors are convinced that RSV-A was not the cause of the exacerbation. From the present study's data it may be hypothesised that low viral loads of RSV-A may have facilitated infection with other respiratory pathogens. However, the present study was not designed to investigate latent RSV-A infection in COPD, as the analysis of several samples taken at different time points, in a prospective longitudinal study, would be needed.
The difference between the correspondence of results of cell and cell-free samples of the ARI (100%) and COPD group (17%) may also indicate different roles of RSV in these diseases. High viral load in acute infection always seems to result in cell lysis whereas in chronic infection, with low viral load, this may be not the case.
In summary the authors' respiratory syncytial virus-A specific quantitative real-time polymerase chain reaction assay is a specific, sensitive, reliable and rapid method for application for the medical diagnosis and quantification of the respiratory syncytial virus genome in clinical samples. It has several advantages compared to traditional methods of viral detection and to other polymerase chain reaction technologies. Theexact determination of viral load allows differentiated interpretation of results and further access to study the role of respiratory syncytial virus in respiratory tract diseases. As an example the authors demonstrate that the different roles of respiratory syncytial virus infection in children compared to adults with chronic obstructive pulmonary disease can be detected by a quantitative real-time polymerase chain reaction assay. Further studies will show whether this is specific for respiratory syncytial virus infection or can also be found with other viruses.
Acknowledgments
The authors would like to thank U. Arinir, A. Wiethege, A. Schürmann and P. Keller for clinical management of patients and recruiting the chronic obstructive pulmonary disease study group and especially M. Kauth for helpful discussions. The authors are also grateful to E. Dretaki-Schnackenberg and S. Werner for their excellent technical assistance (all from the University Hospital Bergmannsheil, Dept of Internal Medicine, Division of Pneumology, Allergology and Sleep Medicine, Germany).
- Received September 24, 2002.
- Accepted January 29, 2003.
- © ERS Journals Ltd
References









