





Abstract
The aim was to investigate whether microRNA (miRNA) expression is modulated by inhaled corticosteroid (ICS) treatment
We performed genome-wide miRNA analysis on bronchial biopsies of 69 moderate/severe chronic obstructive pulmonary disease (COPD) patients at baseline and after 6- and 30-month treatment with the ICS fluticasone propionate or placebo. The effect of ICS on miRNA expression was validated in differentiated primary bronchial epithelial cultures, and functional studies were conducted in BEAS-2B cells. MiRNAs affected by ICS and their predicted targets were compared to an independent miRNA dataset of bronchial brushings from COPD patients and healthy controls.
Treatment with ICS for both 6 and 30 months significantly altered the expression of four miRNAs, including miR-320d, which was increased during ICS treatment compared with placebo. The ICS-induced increase of miR-320d was confirmed in primary airway epithelial cells. MiR-320d negatively correlated targets were enriched for pro-inflammatory genes and were increased in the bronchial brushes of patients with lower lung function in the independent dataset. Overexpression of miR-320d in BEAS-2B cells dampened cigarette smoke extract-induced pro-inflammatory activity via inhibition of nuclear factor-κB.
Collectively, we identified miR-320d as a novel mediator of ICS, regulating the pro-inflammatory response of the airway epithelium.
Abstract
MiR-320d is a novel mediator of inhaled corticosteroids, regulating the pro-inflammatory response of the airway epithelium http://ow.ly/r4IV30nCQeE
Introduction
Chronic obstructive pulmonary disease (COPD) is characterised by chronic, not fully reversible airflow limitation that is usually progressive and associated with an inflammatory response in the airways [1]. Inhalation of noxious gases, including cigarette smoke, is the major risk factor for COPD. Several studies have investigated the efficacy of inhaled corticosteroids (ICS) in COPD [2–4], with conflicting results [2–4]. Only a subgroup of patients responds well to ICS. Given this heterogeneity, it is of relevance to perform studies on therapy response in COPD. Our Groningen and Leiden Universities study of Corticosteroids in Obstructive Lung Disease (GLUCOLD), a double-blind, randomised, placebo-controlled study to investigate the long-term efficacy of fluticasone propionate with or without added salmeterol in COPD, found a greater improvement in lung function decline upon ICS treatment than most earlier studies [5]. This may have been due to the distinct inclusion criteria, requiring patients to not have had ICS treatment for ≥6 months before inclusion in the study, and 95% of patients never used ICS at all. In GLUCOLD, placebo-treated patients experienced a considerable decline in their forced expiratory volume in 1 s (FEV1), whereas the rate of FEV1 decline was close to zero for fluticasone propionate and fluticasone propionate/salmeterol treatment [5]. These results suggest that ICS can be beneficial, at least in a subgroup of COPD patients. In a follow-up study, we performed genome-wide gene-expression analysis on bronchial biopsies from GLUCOLD patients at baseline, and after 6 and 30 months of treatment [6], which enabled us to distinguish between ICS-sensitive and -insensitive patients based on their gene-expression profiles. However, the mechanisms controlling ICS-induced gene-expression changes remained unknown, and understanding this process may lead to identification of novel anti-inflammatory pathways.
MicroRNAs (miRNA)s are small noncoding messenger (m)RNA transcripts with the ability to degrade or inhibit mRNA transcripts, leading to suppression of protein levels. Since each individual miRNA can modulate the expression of >100 genes [7], variations in miRNA levels can radically alter gene-expression profiles. In the present study, we hypothesised that miRNAs are crucial in regulating ICS-induced gene expression in the airways of COPD patients. To investigate this, we assessed alterations in miRNA expression after short- and long-term treatment with ICS, and we were able uncover their target genes, since both miRNA and mRNA data were available from the same bronchial biopsies. We confirmed the functional role of the identified miRNAs in vitro in human bronchial epithelial cells. Our study identified miR-320d, as a novel anti-inflammatory miRNA that is upregulated by corticosteroids.
Methods
Patients and study design
MiRNA expression profiling was performed in bronchial biopsies from COPD patients participating in the GLUCOLD study [5]. The inclusion criteria and GLUCOLD study design have been described previously [5]. Briefly, patients were required to be without ICS treatment for ≥6 months before inclusion, current or ex-smokers with moderate to severe COPD. Patients included in this study were treated with 1) placebo twice daily for 30 months; 2) fluticasone propionate 500 µg twice daily for 30 months; or 3) fluticasone propionate 500 µg + salmeterol 50 µg twice daily for 30 months (table 1). Bronchial biopsies were taken at baseline and 6 and 30 months after treatment for microarray gene-expression profiling and histology. The study was approved by the local medical ethics committee and all patients provided their written informed consent. Methods for miRNA isolation, labelling, microarray hybridisation (GeneChip miRNA 1.0 Array; Affymetrix, Santa Clara, CA, USA) are described in the supplementary material, while the methods for the mRNA extraction and microarray processing have been described previously [6]. Gene- and miRNA-expression data are available through the Gene Expression Omnibus repository (www.ncbi.nlm.nih.gov/geo/) with the accession numbers GSE36221 and GSE76774, respectively. In an independent cohort, miRNA expression profiling was obtained from bronchial brushings from 30 COPD (defined by FEV1/forced vital capacity <70 and FEV1 <80% predicted) and 30 non-COPD controls using Illumina HiSeq (San Diego, CA, USA).
Patient demographics
Statistics
To identify miRNAs altered by ICS treatment, we performed linear mixed effects models with treatment as a categorical variable with two levels: ICS or ICS + long-acting β2-agonist (LABA) (two separate analyses were run) versus placebo (R statistical software, V3.0.2; www.r-project.org). Time was defined as a categorical variable with three levels: baseline, 6- and 30-month treatment. These methods are explained in extensive detail in the supplementary material.
Predicted target approach
To identify the potential targets of differently expressed miRNAs, treatment-induced changes in miRNA expression were compared to changes in mRNA expression in the same biopsies [6]. Pearson correlations were conducted at 1) baseline and 2) change in miRNA and mRNA expression after 6 months focusing on predicted mRNA targets for each miRNA (based on the combination of Target Scan v7.1 [7] and DIANAmicroT). Negatively correlated miRNA targets that were negatively correlated with both analyses (p<0.05) were selected for pathway analysis using Gene Set Enrichment Analysis (GSEA) and GeneNetwork [6].
Following this analysis, the negatively correlated predicted targets altered by ICS were studied in bronchial brushings from 63 smokers with COPD and 135 control smokers without a history of using ICS or oral corticosteroids in an independent cohort using the continuous COPD-related measurement of FEV1. GeneNetwork analysis was performed to investigate the predicted targets of identified miRNAs (nominal p-value <0.05) [8].
GeneNetwork analysis uses an independent gene expression dataset of ∼78 000 samples to predict the function of genes in an unbiased way. We used this method to predict (currently unknown) gene functions based on known biological pathways available in the molecular signatures database MSigDB (GO biological process and the reactome) (www.broadinstitute.org) [8].
Primary bronchial epithelial cell culture
Primary bronchial epithelial cells (PBECs) were obtained from tracheobronchial tissue of nine healthy lung donors by pronase treatment as described previously [9]. Cells were cultured as described previously [9] and as detailed in the supplementary material. MiRNA expression was assessed by real-time PCR as described in detail in the supplementary material.
Preparation of cigarette smoke extract
Cigarette smoke extract (CSE) was prepared as described previously and in the supplementary material [9]. Concentrations of CSE used for each cell line was evaluated by a concentration series of 0–50% CSE and then annexin V and propidium iodide (PI) staining [10]. A concentration for each cell line was selected to minimise cellular death compared to control. For BEAS-2B 10% CSE was selected while for PBECs 20% CSE was selected (supplementary figure S1A and B).
MiR-320d overexpression in BEAS-2B cells and PBECs
Human bronchial epithelial cell line BEAS-2B and PBECs were cultured as described previously [11]. For transfection, cells were seeded in duplicates in RPMI/5% fetal bovine serum or bronchial epithelial cell growth medium at 1×105 in 24-well plates for BEAS-2B and PBEC, respectively. After 24 h, cells were then transfected with miR-320d mimic (1 nM; Qiagen, Venlo, the Netherlands) and nontargeting control (1 nM; Qiagen) using RNAimax (Invitrogen, Groningen, the Netherlands), grown to confluence, serum-deprived for 24 h and treated with BEAS-2B (10% CSE for 24 h in presence and absence of fluticasone propionate (10−8 M) pre-treatment for 3 h) while PBECs were treated with 20% CSE for 8 and 24 h. Annexin V PI staining was used to confirm that no additional cellular death occurred between the transfection of scrambled and miR-320d mimic (supplementary figure S1C). A higher dose of CSE was selected for PBECs, as we have shown previous that primary cells are less sensitive to CSE than cell lines [12]. Cell lysates and cell-free supernatants were collected to determine the miR-320d expression and CXCL8 levels at the 24 h time point (R&D Systems, Abingdon, UK), while mRNA was collected at 8 h stimulation for PBECs and extracted to measure interleukin (IL)-1β and CXCL8 mRNA levels using quantitative PCR [13].
Gene-expression analysis in publicly available datasets
To investigate genes which lead to an increase of CXCL8 during smoke exposure we analysed a publicly available dataset (GSE30660) of air–liquid interface (ALI)-differentiated PBECs (n=4) from healthy donors with/without repeated cigarette smoke challenge (30 min exposure on four separate days). We focused on genes that were increased by cigarette smoke (fold change >2, false discovery rate (FDR) <0.05) using R version 3.1.4 and the limma package.
Nuclear factor-κB activity
To investigate whether overexpression of miR-320d directly suppresses nuclear factor (NF)-κB activity, we used a renilla luciferase reporter assay according to the manufacturer's instructions (Promega, Leiden, the Netherlands) as described in the supplementary material.
Results
Change in miRNA expression after 6- and 30-month treatment with ICS
We identified six miRNAs with altered expression after 6 months of ICS treatment compared with placebo in bronchial biopsies of COPD patients (FDR <0.25). A full list is presented in table 2 and a heatmap and volcano plots are illustrated in figure 1a–c. Four of these six miRNAs (downregulated: miR-708 and miR-155; upregulated: miR-320d and miR-339-3p) remained significant in the same direction after 30 months treatment (nominal p<0.05). None of the four miRNAs identified were found to be altered between current and ex-smokers at baseline, indicating that smoking has no effect on these miRNAs (data not shown).
MicroRNAs (miRNAs) that changed after 6 and 30 months of inhaled corticosteroid treatment compared with placebo

MicroRNA (miRNA) altered by inhaled corticosteroid (ICS) treatment compared to placebo in bronchial biopsies of chronic obstructive pulmonary disease (COPD) patients. a, b) Heatmap showing changes in miRNA expression in response to ICS versus placebo at a) 6 and b) 30 months compared to baseline in bronchial biopsies of COPD patients; c) volcano plot showing changes in miRNA expression in response to ICS versus placebo after 6 months; d) cluster diagram of miRNA altered by corticosteroids and their anti-correlated predicted targets. Blue text indicates miRNAs decreased by ICS, while red text indicates miRNAs increased by ICS; blue nodes indicate genes decreased by 6 months of ICS treatment compared to placebo, red nodes indicate gene increased by ICS treatment and black nodes indicate no change during ICS treatment; the thickness of the edges (lines connecting miRNAs and genes) is associated with the strength of correlations between miRNA and target messenger RNA.
Associations between changes in miRNA expression and mRNA expression
We next examined whether the miRNAs altered by ICS negatively regulate the expression of their predicted targets in the COPD-derived bronchial biopsies. We conducted a cross-sectional analysis at baseline and longitudinal analysis between 6 months and baseline. All four identified miRNAs had at least one significant negative-correlated predicted target within either the cross-sectional or longitudinal analysis, with an overlap between these analyses for each miRNA ranging between 30.9% and 80.7% (supplementary figure S2A–D). Supplementary table S1 presents all associations. Connected network analysis identified two main clusters of miRNAs and their predicted targets: 1) miR-708 and miR-155, which were decreased by ICS treatment; and 2) miR-320d and miR-339-3p, which were increased by ICS treatment (figure 1d).
Replication of miRNA altered by ICS
For further validation of the identified miRNA, we investigated the third arm of the GLUCOLD cohort, i.e. patients treated with ICS + LABA. A candidate-based approach identified that of the four miRNAs, miR-320d, miR-339-3p and miR-708 were also significantly altered in the same direction by ICS + LABA compared to placebo at 6 months treatment (p<0.05). A comparison between the ICS and ICS + LABA analysis is illustrated in supplementary figure S3 Next, we investigated in the independent dataset whether miR-320d, miR-339-3p and miR-708 were altered between COPD patients using ICS (n=6) compared to COPD patients not on ICS (n=24). For the replication, we focused additionally on the miR-320 family, as miR-320a-d all have very similar sequences and have the same predicted targets (supplementary figure S4A). Here we found that the miR-320 family (miR-320a, miR-320b-1, miR-320b-2 and miR-320c-2) were significantly increased by ICS, while miR-320c-1 and miR-320d was found to have a trend for an increase (p=0.057 and p=0.086, respectively) reflecting the results found in initial analysis (supplementary figure S4B–G and supplementary table S2). MiR-339-3p and miR-708 were not found to be significantly altered by ICS in the same direction.
Pathway analysis of miRNA targets
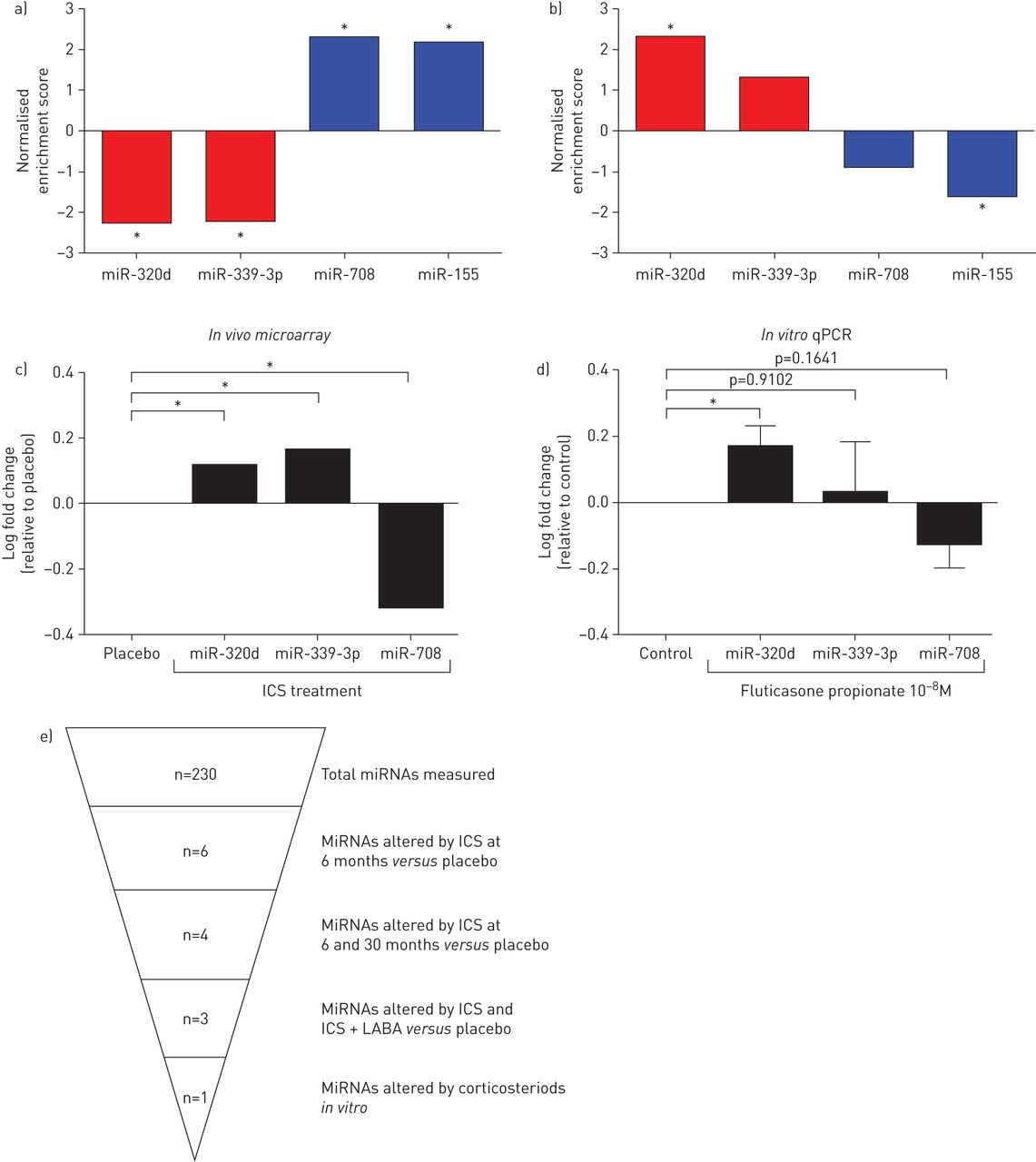
Using GeneNetworks, we found that miR-320d negatively correlated predicted targets were associated with pro-inflammatory related pathways including tumour necrosis factor (TNF)-α signalling and cytokine production, while miR-339-3p predicted targets were associated with regulation of ion transport (nominal p-value <0.05). Finally, miR-708 negatively correlated predicted targets were found to associate with the regulation of ion transport and activation of phospholipase C activity (nominal p-value <0.05). The list of the top five pathways for each miRNA is displayed in table 3; an extended list is provided in the supplementary material (supplementary table S4). To evaluate whether the miRNA negatively correlated predicted targets were ICS sensitive, we performed GSEA on the GLUCOLD data before and after 6 months of ICS treatment compared to placebo. As expected, the majority of miRNA negatively correlated predicted targets were altered by ICS in the opposite direction to their targeting miRNA, with miR-708 negatively correlated targets being increased and miR-339-3p and miR-320d negatively correlated targets being decreased by ICS (figure 2a and supplementary figure S5).
Top five pathways associated with predicted microRNA gene targets

Gene Set Enrichment Analysis of microRNA (miRNA) negatively correlated predicted targets and in vitro validation. a) Negatively correlated predicted targets of miR-320d and miR-339-3p were significantly decreased following inhaled corticosteroid (ICS) treatment compared to placebo in bronchial biopsies (6 months – baseline), while miR-708 predicted targets were increased by ICS treatment; b) negatively correlated predicted targets altered by ICS of miR-320d were increased in chronic obstructive pulmonary disease (COPD) bronchial brushings compared to non-COPD, while miR-708 predicted targets were decreased in COPD; c) average log fold change in miRNA expression upon treatment with ICS compared to placebo at 6 months of treatment; d) log fold change of miRNA expression in epithelial cells grown at the air–liquid interface and treated for 24 h with fluticasone (10−8 M) compared to untreated control. Data are presented as mean±sem (n=9). Paired t-tests were used to test for statistical significance; e) graphical overview of miRNA results. qPCR: quantitative PCR; LABA: long-acting β2-agonist. *: p<0.05.
Treatment-related changes in miRNA expression are associated with presence of COPD or severity of airflow obstruction in an independent cohort
To determine whether miRNAs affected by ICS treatment are clinically relevant, we investigated the association of miRNA expression and presence of COPD and level of FEV1 % pred in an independent cohort of bronchial brushings, where miRNA expression profiles were available from 30 smokers with and 30 without COPD [14]. Of the three miRNAs altered following ICS treatment, miR-708 was found to be altered in the opposite direction between COPD and controls, with higher expression in COPD-derived bronchial brushes (p<0.05). No difference was found for miR-320d or miR-339-3p.
Next, to investigate whether the negatively correlated miRNA predicted targets are relevant to disease pathogenesis, we performed GSEA on an expanded population from the same independent cohort with whole-genome gene-expression data available in bronchial brushings of 63 current or former smokers with and 135 without COPD [14]. As with the miRNA analysis, it was conducted on the continuous COPD-related FEV1 measurement. The miR-320d negatively correlated predicted targets were enriched among the genes increased in subjects with lower lung function, while the miR-708 negatively correlated predicted targets were enriched among the genes decreased in subjects with lower lung function (figure 2b and supplementary figure S6). No such association was found for miR-339-3p predicted targets.
MiR-320d is upregulated by corticosteroid treatment in vitro
Next, we examined whether ICS directly regulate the expression of the identified miRNAs in ALI-differentiated PBECs from healthy donors. Treatment with fluticasone propionate (10 nM) for 24 h resulted in significant upregulation of miR-320d, while miR-708 expression trended to a decrease and miR-339 was not altered (figure 2c and d).
MiR-320d acts as an anti-inflammatory miRNA through regulation of IL-1β-induced NF-κB signalling
As miR-320d was predicted to target genes associated with cytokine production, we hypothesised that miR-320d regulates the expression of pro-inflammatory cytokines/chemokines such as CXCL8, a major pro-inflammatory cytokine that is increased in COPD. To investigate this, we overexpressed miR-320d in the human bronchial epithelial cell line BEAS-2B and PBECs (figure 3a) and assessed CXCL8 release in response to 10% and 20% CSE. Overexpression of miR-320d significantly reduced CSE-induced CXCL8 release in BEAS-2B and PBECs (figure 3b and c). In addition, overexpression of miR-320d reduced CXCL8 mRNA levels during smoke exposure in the PBECs, supporting the protein data (supplementary figure S7A). Incidentally, we found that smoking has a minimal effect on CXCL8 mRNA levels at 6 h. CXCL8 is not a predicted target gene of miR-320d and therefore other upstream mediators are probably involved. To investigate how miR-320d affects cigarette smoke-induced pro-inflammatory epithelial responses, we used a publicly available dataset of ALI-cultured PBECs derived from healthy controls that were either exposed or not exposed to gaseous cigarette smoke (GSE30660). 62 genes were increased by cigarette smoke treatment (fold change >2, FDR <0.05). String network analysis identified that these 62 genes had an enrichment for protein–protein interactions and that this list was associated with chronic inflammation (supplementary table S4, supplementary figure S7B). Of interest, IL-1β was identified as a hub gene in this network (supplementary figure S7C and D); IL-1β is a cytokine previously identified as a key positive regulator of inflammation in COPD increased by cigarette smoke exposure [15]. We previously observed that CSE upregulates IL-1 mRNA expression in PBECs [16]. Furthermore, we found that overexpression of miR-320d at baseline significantly decreases IL-1β mRNA levels (supplementary figure S7E). These results suggest that CSE induces CXCL8 production by activating the IL-1β pathway.

{kind=link}
{kind=link}
{kind=link}
Functional analysis of miR-320d. BEAS-2B and primary bronchial epithelial cells (PBECs) were grown to confluence and serum-deprived overnight. a) Nontargeting microRNA (miRNA)/mimic control or miR-320d was overexpressed in the BEAS-2B and PBECs and its expression was determined using quantitative PCR; b, c) CXCL8 secretion upon overexpression of miR-320d mimic and mimic control in response to 24-h exposure to cigarette smoke extract (CSE) (n=6) in b) BEAS-2B and c) PBECs; d) nuclear factor (NF)-κB activity assay in BEAS-2B overexpressing miR-320d mimic and mimic control in response to 24-h exposure to interleukin (IL)-1β (n=6). e) Diagram of proposed miR-320d function. Activity was expressed as NF-κB activity compared to untreated scrambled control. Data are presented as mean±sem. Paired t-tests were used to test for statistical significance. *: p<0.05.
Based on these findings, we assessed whether miR-320d can attenuate IL-1β-induced CXCL8 production via inhibition of NF-κB, a well-known downstream mediator of IL-1β pro-inflammatory function. To this end, BEAS-2B cells were transfected with a reporter construct for NF-κB and treated with IL-1β for 24 h in the presence and absence of miR-320d overexpression. IL-1β treatment significantly activated NF-κB signalling, and miR-320d overexpression was found to significantly decrease the IL-1β-induced activation of NF-κB signalling compared to negative miRNA control (figure 3d). A proposed mechanism of miR-320d is provided in figure 3e.
Discussion
In the present study, we identified four miRNAs that are affected by short- and long-term treatment with ICS compared to placebo in patients with moderate to severe COPD. Additionally, we show that the predicted targets of these miRNAs are altered by ICS treatment and that specifically miR-320d targets are enriched among genes associated with decreased lung function. In vitro, we confirmed the direct effect of ICS on miR-320d expression in bronchial epithelium and observed that miR-320d overexpression decreases CSE-induced CXCL8 release. This anti-inflammatory function was found to be mediated in part by inhibition of the IL-1β/NF-κB pathway.
We further observed that the negatively correlated predicted targets of miR-320d are decreased by ICS treatment. Although miRNA profiling in bronchial brushes in an independent cohort showed no significant decrease in miR-320d with COPD, miR-320d predicted target genes were increased with COPD in this cohort. The latter may indicate that miR-320d regulation of gene expression may be disturbed in COPD
We have, for the first time, identified an anti-inflammatory role of miR-320d. This was suggested by our pathway analysis, which identified miR-320d as a negative regulator of genes associated with pro-inflammatory pathways, including TNF-α signalling and cytokine production. The miR-320d anti-inflammatory role was validated in vitro in airway epithelium, since we showed that miR-320d overexpression suppresses CSE-induced CXCL8 release through inhibition of NF-κB activation. Here, IL-1β stimulation was shown to be a potent inducer of NF-κB activity. Our data are in line with previous studies showing that airway epithelial cells release IL-1β in response to cigarette smoke treatment [17, 18]. Genes associated with the NF-κB pathway were upregulated in differentiated PBECs treated with cigarette smoke, and increased NF-κB activity has been observed in COPD [19, 20]. CXCL8 is a key mediator in neutrophil infiltration into airway tissue acting as neutrophil chemoattractant [21].
Cigarette smoke provides one of the main inflammatory initiators in COPD [22, 23]; however, the mechanism responsible for aberrant inflammatory response in smokers with COPD and why this cannot efficiently be suppressed by ICS in all patients remains uncertain. Our results support the hypothesis that miR-320d regulates cigarette smoke-induced CXCL8 by inhibiting NF-κB signalling, and may thus be involved in the anti-inflammatory effect of ICS.
In addition, miR-155 may be of potential interest in regulating inflammatory responses, as it was identified in our original analysis and it has previously been shown to be associated with airway inflammation in murine models of asthma [24]. However, we did not investigate miR-155 further as we could not replicate its downregulation in our second analysis.
In contrast to our findings, a previous cross-sectional study was unable to find differences in the miRNA expression profile in bronchial biopsies between treatment with and without ICS in asthmatics [25]. We believe that the difference between the studies may be due to longitudinal nature, rigorous selection criteria and higher power of the current study, or possibly the inherent difference between asthma and COPD patients.
One of the main strengths of this study is the co-analysis of global miRNA and gene-expression change over time, therefore allowing us to correlate changes in miRNA expression with longitudinal changes in gene expression of predicted miRNA targets. This experimental setup overcomes one of the main unknown assumptions of cross-sectional studies on matched miRNA and mRNA samples, namely that the relationship between miRNA and gene expression remains consistent over time [26, 27]. Validation of our finding in an in vivo mouse model of COPD may provide further insight into the anti-inflammatory role of miR-320d [28].
There are some limitations to our study. Despite all samples being snap frozen at −80°C following collection, RNA degradation was found in some samples, as reflected by relatively low RNA integrity number (RIN) scores. Therefore, we adjusted for RIN score values in all analyses conducted in this study. Bronchial biopsies contain a mixture of cell populations. Importantly, after adjusting for inflammatory cell counts, miR-320d, miR-339-3p and miR-708 remained significant, indicating that the change in miRNA expression was not due to an ICS-induced change in inflammatory cell populations. Finally, as we used miRNA arrays we may have missed a number of novel miRNAs.
In summary, we have profiled global miRNA expression in bronchial biopsies from patients with moderate to severe COPD, following longitudinal treatment with ICS compared to placebo at baseline, 6 and 30 months. We found four miRNAs (miR-320d, miR-339-3p, miR-708 and miR-155) to be altered following ICS treatment. Furthermore, we identified miR-320d as a novel anti-inflammatory miRNA that suppresses NF-κB activity in vitro. MiRNAs associated with ICS treatment and inflammation provide important candidates for future studies as possible biomarkers and therapeutic targets in chronic inflammatory diseases.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-01202-2018.Supplement
Supplementary table S1 ERJ-01202-2018.Table_S1
Footnotes
This article has supplementary material available from erj.ersjournals.com
Author contributions: A. Faiz performed microarray analysis, in vitro cellular work, project design and writing of the manuscript. S.D. Pouwels, M.P. Roffel, T.R. Eijgenraam and M. Borggrewe aided with the in vitro cellular work. D.S. Postma, I.H. Heijink, A. Spira and G.H. Koppelman participated in project design and drafting of the manuscript. I.H. Heijink supervised the practical work. S. Lam collection of non-COPD and COPD bronchial brushings. K. Steiling performed the analyses comparing microRNA expression in non-COPD and COPD and writing of the manuscript. M. van den Berge performed microarray analysis, project design, and writing of the manuscript. M.R. Jonker contributed to the in vitro cellular work. P.S. Hiemstra, P.J. Sterk and W. Timens participated in the revision of the manuscript. C-A. Brandsma performed the pathway analysis. G. Liu and Y.O. Alekseyev processed mRNA and miRNA samples for microarray analysis. All authors read and critically reviewed the manuscript.
Conflict of interest: A. Faiz has nothing to disclose.
Conflict of interest: K. Steiling has a patent “Biomarkers of COPD disease activity” pending.
Conflict of interest: M.P. Roffel has nothing to disclose.
Conflict of interest: D.S. Postma reports institutional grants from AstraZeneca, Chiesi, Genentec, GSK and Roche, and fees for consultancy from Astra Zeneca, Boehringer Ingelheim, Chiesi, GSK, Takeda and TEVA.
Conflict of interest: A. Spira has nothing to disclose.
Conflict of interest: M.E. Lenburg has nothing to disclose.
Conflict of interest: M. Borggrewe has nothing to disclose.
Conflict of interest: T.R. Eijgenraam has nothing to disclose.
Conflict of interest: M.R. Jonker has nothing to disclose.
Conflict of interest: G.H. Koppelman reports institutional grants from Netherlands Lung Foundation, Ubbo Emmius Foundation, TEVA Pharmaceuticals and Stichting Astma Bestrijding, outside the submitted work
Conflict of interest: S.D. Pouwels has nothing to disclose.
Conflict of interest: G. Liu has nothing to disclose.
Conflict of interest: Y.O. Alekseyev has nothing to disclose.
Conflict of interest: S. Lam has nothing to disclose.
Conflict of interest: P.S. Hiemstra reports grants from Netherlands Organization for Scientific Research, Netherlands Asthma Foundation, University of Groningen, Leiden University Medical Center and GlaxoSmithKline, during the conduct of the study.
Conflict of interest: P.J. Sterk reports grants from GlaxoSmithKline and the Dutch Government, during the conduct of the study.
Conflict of interest: W. Timens reports personal fees for consultancy from Pfizer and Merck Sharp Dohme, personal fees for lecturing from GSK, Lilly Oncology and Chiesi, personal fees for lecturing, consultancy and travel from Roche Diagnostics/Ventana, travel costs from Biotest, personal fees for lecturing and consultancy Novartis, and grants from the Dutch Asthma Fund, outside the submitted work.
Conflict of interest: C-A. Brandsma has nothing to disclose.
Conflict of interest: I.H. Heijink has nothing to disclose.
Conflict of interest: M. van den Berge reports institutional research grants from Chiesi, Teva Pharma and AstraZeneca, outside the submitted work.
Support statement: This work was supported by the Longfonds, Netherlands, Junior Investigator grant (number 4.2.16.132JO; A.Faiz) and European Respiratory Society RESPIRE2 fellowship.
- Received January 24, 2018.
- Accepted January 30, 2019.
- Copyright ©ERS 2019
References










