Article Text

Abstract
Epigenetic mechanisms are believed to play an important role in disease, development and ageing with early life representing a window of particular epigenomic plasticity. The knowledge upon which these claims are based is beginning to expand. This review summarises evidence pointing to the determinants of epigenetic patterns, their juxtaposition at the interface of the environment, their influence on gene function and the relevance of this information to child health.
Statistics from Altmetric.com
What is epigenetics?
Waddington1 first described epigenetics, defining it as the study of epigenesis, a field which we now know as developmental biology. Over the last 60 years the definition of epigenetics has evolved and been refined. Bird2 defines epigenetics as “the structural adaptation of chromosomal regions so as to register, signal or perpetuate altered activity states”. The modern definition of epigenetics therefore encapsulates changes to the DNA or associated proteins, excluding changes to the DNA sequence itself while emphasising the role of epigenetic events in ‘capturing’ information about environmental exposures. Importantly, these changes are heritable during cell division.
At the molecular level, there are three groups of epigenetic mechanisms. These comprise methylation (a biochemical alteration of the DNA), modification of the histones which package the DNA, and non-coding or interference RNAs (including microRNAs (miRNAs)) that maintain or change transcription in a heritable way.
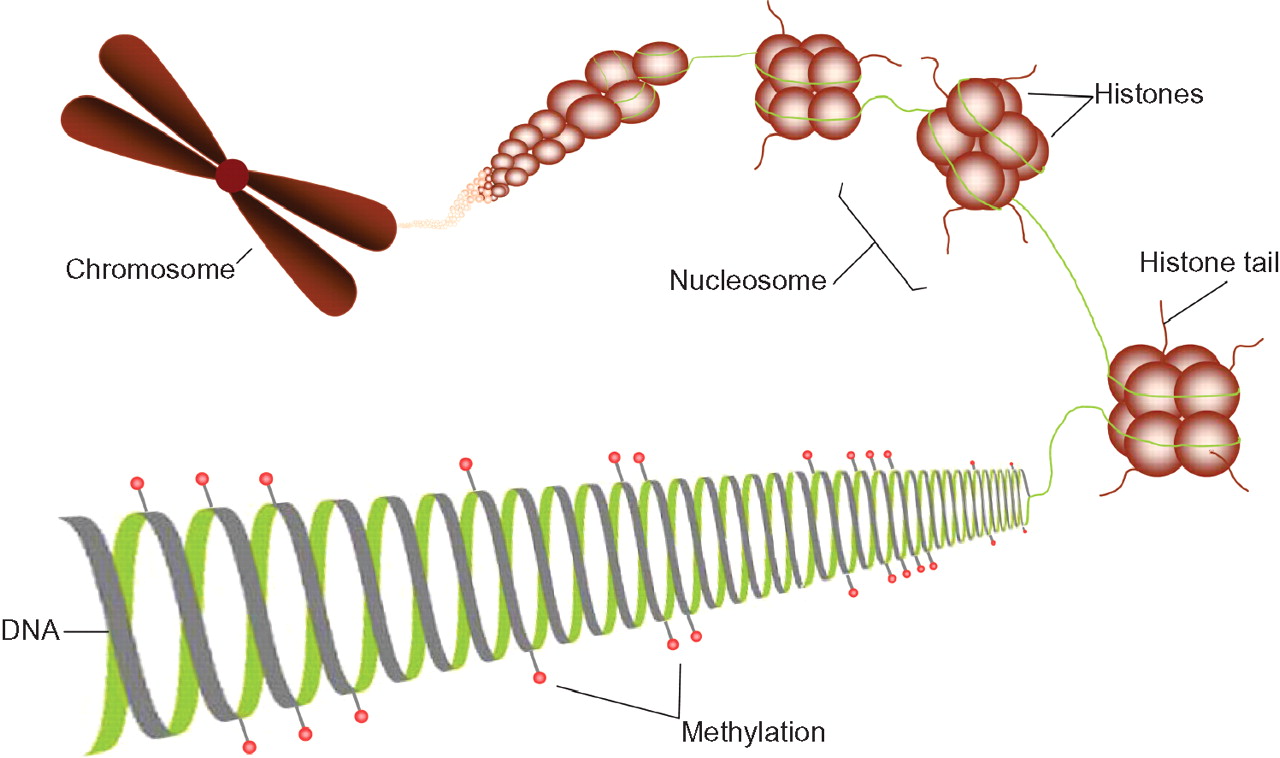
Methylation is the best understood epigenetic process. It involves a biochemical modification of the DNA: a methyl group is bound to the carbon-5 position of a cytosine base, in the presence of DNA methyltransferases (1, 2, 3a and 3b) to form 5-methylcytosine. In mammals, this addition occurs only where a cytosine is adjacent to a guanine residue. These locations are referred to as CpGs, the ‘p’ indicating the phosphate group between the di-nucleotide pair. Approximately 7% of the cytosine bases in the mammalian genome are methylated and methylation is conserved during cell division. DNA methylation and histone modification, as well as other forms of epigenetic modification, do not act independently of one another (see figure 1), for example, methylation of specific regions can act to recruit histone deacetylases leading to gene silencing.3 4

The epigenetic code. Methylation of DNA at CpG sites, indicated by red ‘lollipops’, is involved in regulating gene expression. Histone tails can be modified by acetylation, methylation, phosphorylation and ubiquitination, which alter the packaging of chromatin. Condensation of the chromatin leads to changes in the accessibility for binding of transcription machinery, and therefore downstream changes in gene regulation.
Histone modifications are also well studied and include acetylation, methylation, phosphorylation and ubiquitination of (primarily) histone tails5 6 which allow the chromatin to become more or less condensed. Condensation of the chromatin leads to changes in the availability for binding of transcription machinery, and therefore downstream changes in gene regulation. Unlike methylation, the mode of transmission of histone modification from parent to daughter cell is less well understood.
miRNAs are small, non-coding, single stranded RNA molecules which are able to downregulate gene expression through a number of pathways. The most common mechanism in mammals is by translational repression, where a miRNA binds to a messenger RNA molecule causing reduced protein expression downstream.7 miRNAs have been shown to be capable of regulating epigenetic pathways, targeting enzymes involved in both histone modification8 9 and methylation.10
What is the purpose of epigenetic changes?
Epigenetic marks are closely tied to the control of gene expression and ultimately function. In the case of methylation, CpG sites are found throughout the genome but at a lower than expected frequency. Conversely, some regions of the genome, especially promoter regions of genes, are frequently rich in CpGs; these regions are known as CpG islands. Methylation of CpG sites within these islands is very important in terms of transcription and regulation of gene expression as methylation of CpG sites in these regions may alter transcription factor binding mechanisms or lead to recruitment of co-repressors.11
Epigenetic mechanisms have been studied in detail in relation to early development due to their role in cell differentiation.12 In early embryogenesis two phases of epigenetic reprogramming occur: a widespread demethylation of the genome followed by de novo methylation.12 If perturbations of this process occur, the resultant methylation in the many cell lineages of the offspring may be altered. A second focus of epigenetic research has been in the field of oncology as epigenetic perturbation is a common feature of tumours. More recently, the juxtaposition of epigenetic variation at the interface between the genes and the environment has generated increasing levels of interest in its role in many other common complex diseases including type II diabetes, cardiovascular disease and autism spectrum disorders (ASD).13 14
The epigenome ‘memorises’ environmental signals
The methylation status of any given CpG site is not fixed, and thus the epigenome can be thought of as a fluid system. This is bi-directional, and methylation and demethylation may occur. The ability of the individual to both methylate their genome and to maintain those methylation patterns through multiple cell divisions are important concepts when considering epigenetic fidelity and stochastic influences on epigenetic patterns.
Environmental cues can lead to changes in methylation in differentiated somatic tissues and thus epigenetics can be thought of as the interface between genes and environment. Changes in methylation status occurring over a person's lifetime have been associated with ageing. Current evidence supports a generalised loss of fidelity in epigenetic patterns with age, confined mainly to repeat, non-coding sequences within the genome.14 As yet, there is a dearth of evidence linking epigenetic variation to age-related phenotypic traits, although this may be due largely to the lack of research undertaken in this field to date. More importantly, for child health, there is evidence that the epigenome is particularly plastic during early stages of development in utero and early postnatal life. Additionally, recent debate has also focused on the mechanisms underlying the trans-generational transmission of environmentally acquired epigenetic patterns from parents to their offspring.
The neo-Lamarkian concept of a cellular memory of environmental exposure with potential consequences for adaptive response is highly attractive in explaining the rapid transitions and inter-generational amplification observed in health problems, including the recent epidemic of childhood obesity. Care must however be taken when interpreting epigenetic inheritance, as mitotic inheritance of epigenetic markings is distinctly different to meiotic inheritance. The epigenome undergoes comprehensive re-modelling postfertilisation and during primordial germ cell development,12 wiping clean acquired epigenetic patterns – or so current dogma suggests. Imprinted regions of the genome are protected from demethylation and one might postulate that demethylation may show some degree of inter-individual variation. This has yet to be established. There is some evidence to show that trans-generational inheritance of epigenetic patterns may in part be mediated by the transmission of small interfering RNA species. Trans-generational inheritance of epigenetic patterns has recently been extensively reviewed15 16 and although there is some epidemiological evidence to support this concept,17 there remain many fundamental questions to be answered.
Determinants of epigenetic variation
Methylation status is modulated by several factors; we summarise a selection from the literature here.
Nutrition
Nutritional intake is a well documented environmental modifier of methylation. Low intake of methionine leads to changes in methylation in mice18 and humans. Conversely, an increased intake of folic acid, vitamin B12, choline and betaine during pregnancy lead to increased methylation in offspring in mice. In the Agouti mouse model this increase in methylation leads to altered expression of a coat colour gene, producing different coat colours in the offspring.19 In addition to methyl donors, several other dietary factors influence epigenetic patterns. These include polyphenols, isothiocyanates, genistein, selenium and butyrate, which mediate effects independently of folate metabolism.20 21
Famine during the prenatal period, best exemplified by studies on the Dutch hunger winter families,22 leads to changes in methylation in the IGF2 gene which persist into adulthood.
Increasing evidence has highlighted the potential role of epigenetic mechanisms in the aetiology of fetal alcohol spectrum disorders (FASD). Ethanol consumption has been shown to affect a number of DNA methylation pathways including reduced expression of DNMT3b mRNA23 and elevated levels of homocysteine,24 an important player in one-carbon metabolism. The timing of alcohol consumption may also influence the role of epigenetic mechanisms in FASD risk such as during the preconception and preimplantation periods or during central nervous system development.25
Age
Epigenetic changes due to environmental cues and due to stochastic events accumulate during a person's lifetime. Older monozygotic twins differ more in their amounts of DNA methylation compared to younger twins,26 although such inter-individual variation has not been reported in smaller studies of unrelated individuals.27 In a study of subjects aged 43–70 years selected from individuals exposed prenatally to the Dutch hunger winter of 1944, a 3.6% decrease in IGF2 methylation was associated with each 10-year increase in age.22 Furthermore, a study looking at families in Iceland and the USA demonstrated that over time (11–16 years duration) intra-individual global DNA methylation levels changed, and importantly familial clustering of these changes was also observed.28
Genetic factors
Genetic factors may also play a role in epigenetic mechanisms. Recent evidence reports the heritability of DNA methylation patterns and points to the influence of genetic variation in close proximity to CpG sites influencing methylation (cis effects) as well as more distally positioned genetic variation having an influence of methylation at specific CpG sites (trans effects). CpG island methylation has been shown to be highly correlated with specific DNA repeats, DNA sequence patterns and the predicted DNA structure.29 Leng et al30 reported that hypermethylation of gene promoters in sputum occurs in smokers, and that methylation is associated with a number of single nucleotide polymorphisms in genes involved in DNA repair mechanisms.
Stress
Studies of stress indicate that maternal care may play a role in epigenetic changes. In rats, methylation of the glucocorticoid receptor is reduced in offspring with lower levels of nurturing, leading to increased expression of the receptor in later life.31 This process may also be dynamic, and importantly, reversible.32 In humans, the glucocorticoid receptor gene NR3C1 is also subject to perturbed methylation in response to stress.33 In this study, maternal mood in the third trimester of pregnancy was associated with methylation of NR3C1 and salivary cortisol in offspring at 3 months of age.
Smoking
Methylation is associated with smoking.34 35 Furthermore, methylation levels in sputum may be a valuable biomarker in identifying lung cancer.30 A decrease in global methylation levels has also been demonstrated in children exposed to maternal smoking in utero,36 with differences in global methylation (LINE1) being associated with GSTM1 genotype, the latter being involved in the detoxification of the products of cigarette smoke. Our unpublished findings support the observation that the effects of maternal smoking during pregnancy are discernable in the neonatal epigenome in the form of differential DNA methylation.
Infection
The involvement of maternal or neonatal infection in establishing or altering epigenetic patterns is largely unexplored. A number of studies have looked at the relationship between Helicobacter pylori infection, methylation and gastric cancers. Infection by H pylori increases the risk of developing gastric cancers37 38 and in healthy volunteers has also been shown to cause aberrant CpG hypermethylation of specific genes in gastric mucosae.39
Disease status
Mutations in the epigenome have been shown to give rise to a number of syndromes, including those associated with genomic imprinting, and have also been implicated in ASD. Genomic imprinting occurs where, through epigenetic mechanisms, genes are expressed in a parent of origin manner. It is thought that this mechanism may have evolved to regulate the resources available to the offspring.
Beckwith–Wiedemann syndrome (BWS) is an overgrowth disorder associated with an increased risk of developing Wilms tumour. The epimutation for this syndrome occurs on the maternal allele of chromosome 11p15. There are two differentially methylated regions (DMRs) at 11p15. DMR1 regulates the genes IGF2 and H19 and is hypermethylated in a small proportion of BWS patients. The other epimutation occurs at DMR2 which is hypomethylated in over 50% of BWS patients; this region regulates the genes CDKN1C and KCNQ1.40
Conversely, hypomethylation on the paternal allele of DMR1 has been found in the growth retardation disorder Silver–Russell syndrome (SRS), occurring in over half of those with the disorder.41 This syndrome is unique in that it is associated with three imprinted loci located on two different chromosomes. The other mutations shown to occur within this phenotype are maternal duplication of 11p15 and maternal uniparental disomy of chromosome 7.
Transient neonatal diabetes mellitus (TND) has also been shown to be associated with epimutations. The DMR at 6q24 regulates the genes ZAC and HYMA1 and is normally methylated on the maternal allele. In 20% of TND individuals hypomethylation occurs on the maternal allele at this DMR.42
A number of individuals who have features of one of the disorders described above have been identified as having maternal hypomethylation syndrome.43,–,45 Therefore, as well as having the epimutations associated with a particular disorder, they also have maternal hypomethylation at multiple loci.
Although it remains controversial, a number of studies have indicated there is an increased risk of genomic imprinting disorders in children who are conceived by assisted reproductive technologies (ART) (for a comprehensive review see Laprise46 and Owen and Segars47). During both germ cell and preimplantation development, when ARTs are used, DNA methylation patterns are reset. ARTs therefore have the potential to alter epigenetic markings at these stages in development with the majority of evidence suggesting that this alteration occurs as loss of methylation at the maternal allele. Due to the low incidence of imprinting disorders it is difficult to ascertain the relative risk of these disorders following ART. The disorders which have been reported to be increased include BSW, SRS, Angelman syndrome, maternal hypomethylation syndrome and retinoblastoma.
The role of epimutations in ASD is starting to be elucidated.13 Evidence for this is provided by the single gene disorders fragile X syndrome (FXS) and Rett syndrome, which have been shown to be associated with ASD. FXS results from the expansion of a CGG triplet, to more than 200 repeats, in the 5′ untranslated region of the FMR1 gene. Hypermethylation at this region results in the transcriptional silencing of FMR1. The X linked neurodevelopmental disorder Rett syndrome arises from mutations in the methyl-CpG binding protein 2 (MeCP2) gene. MeCP2 has been shown to have a dual role in gene expression, involved in both transcriptional repression as well as activation.48
Increasing evidence suggests that chromosomes 7q and 15q are involved in ASD.7 As regions of genomic imprinting on these chromosomes have been found in close proximity to regions identified in linkage studies, this highlights further the potential role of epigenetic regulation in ASD.
Critical windows in defining epigenetic patterns
Both the intrauterine environment and early postnatal life are critical windows in developmental plasticity. Throughout these periods the individual is highly susceptible to adaptation towards its environment whether this is a response to chemical or social stresses. In a poor intrauterine environment the fetus may enhance postnatal survival by optimising the growth of key body organs to the detriment of other organs and mechanisms.49 Molecular changes that occur as a consequence of early life exposures are thought to be mediated to a greater or lesser extent by persistent changes in gene expression. Increasing evidence suggests that epigenetic mechanisms are involved in this long term programming of gene expression.
Developmental origins of health and disease
Epigenetic mechanisms are a strong contender for explaining the developmental origins of health and disease. Environmental cues during development can influence gene expression levels and may result in long term health consequences. This early life programming has been shown to be associated with critical periods during development and may also be influenced by gender.50
As alluded to earlier, analysis of the IGF2 gene in the Dutch hunger winter cohort showed that those individuals exposed to famine peri-conceptionally had decreased levels of DNA methylation six decades later compared to same sex siblings. However, those exposed during late gestation showed no differences in DNA methylation levels when compared to controls.22 Further studies have extended these findings to other genes involved in metabolic and cardiovascular disease as well as those involved in growth, some of which were sex specific.50 This latter study also reported that while most genes showed altered methylation following peri-conceptional exposure, some genes showed altered methylation following late gestational exposure. As different genes play different roles at varying stages of development, it is possible that they become epigenetically sensitive during different developmental windows. Considerably more work is required to map and to understand the temporal changes in DNA methylation and epigenetic patterns more broadly during prenatal development.
A critical window of exposure has also been highlighted in allergic airway disease in mice.51 Exposure to a diet rich in methyl donors in utero enhanced the severity of allergic airway disease and predisposition was observed to be partially transmitted to subsequent generations. The effect of this diet was limited to exposure during gestation and not during lactation or adulthood. Increased methylation and decreased expression of the Runx3 gene was observed which is involved in the regulation of CD4/CD8 T lymphocyte development.
Integrating epigenetics into population based studies
Measuring methylation in patients or in the general population requires the collection of DNA samples. These samples may be derived from tissue, but DNA derived from peripheral blood or saliva is most accessible. A recent study reporting variation in DNA methylation across multiple tissues in six individuals showed a higher level of variation in methylation patterns between tissues than between individuals.52 Comparison between study populations where methylation patterns are derived from different tissues should therefore consider correlation between tissue types. DNA analysed from various sources has been described in the literature, however different extraction methods, storage conditions and quality of DNA are likely to influence the integrity and long term stability of DNA methylation patterns and may introduce systematic error into laboratory analysis. These factors should therefore be accounted for if comparing samples from different sources.
Unlike genetic association studies where thousands of individuals are often required, DNA methylation can be considered as a ‘phenotype’. This means that such large study populations are not required; epigenetic based studies may therefore be more appropriate in child health research where smaller cohort sizes are more typical.
Where and how to look for epigenetic changes
The methods currently being used to analyse DNA methylation levels have been extensively reviewed elsewhere,53 54 therefore only a brief summary will be given here.
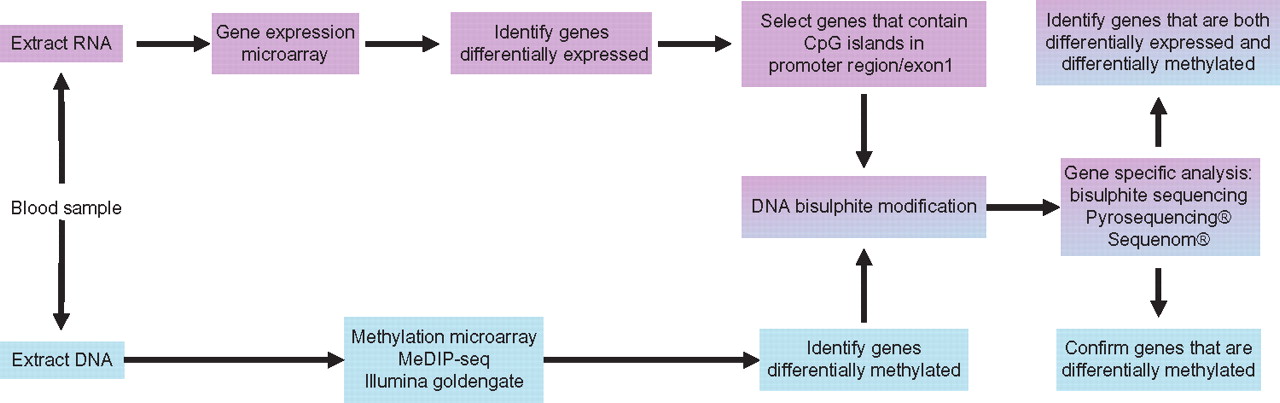
There are two approaches when looking for epigenetic changes within the genome. First, a whole epigenome approach can be taken where the investigator is interested in global changes or is interested in identifying regions of interest from a large dataset. Alternatively, if a putative gene or region of interest is known, a targeted approach to quantifying methylation in a particular region can be adopted. These approaches can be integrated as exemplified by the workflow shown in figure 2.

{kind=link}
{kind=link}
Schematic of workflow to identify genes that are both differentially methylated and differentially expressed in a human cohort. Note current dogmas that DNA methylation of gene regulatory regions is associated with the silencing of gene expression. This relationship is not clear cut, thus restricting interrogation of DNA methylation to only those genes differentially expressed has obvious limitations.
The majority of protocols employed in detecting levels of methylation in DNA samples are based on three methods. The first involves chemical alteration of cytosine residues, known as bisulphite modification, which results in a methylation dependent change in the DNA sequence. This is the most common protocol that analysis systems are currently based upon. Second, there are a number of assays dependent on the enrichment or capture of methylated DNA, for example, methylated DNA immunoprecipitation. The third type of analysis method depends on methylation specific restriction enzymatic digestion. A selection of common methods currently used are shown in table 1.
Summary of methods used to measure methylation from DNA
Relating methylation status to phenotype
Establishing causality presents a continual challenge when linking biomarkers to phenotypic data. The identification of differential methylation in a diseased population when compared to controls could arise from methylation changes secondary to the disease state (reactive model) or the disease may have arisen as a consequence of the methylation changes (causal model). The established relationship between DNA methylation and gene silencing provides the opportunity for corroborative experiments; if altered DNA methylation changes gene expression and gene expression is in turn associated with disease, this would support a causal association.55 In vitro studies using demethylating agents, such as 5-azacytidine, can also be used to confirm the functional importance of DNA methylation changes in target genes.56 57 Longitudinal studies involving the analysis of serial samples from the same individuals at multiple time points will determine not only the direction of causality of methylation changes but also their relative contribution in light of other environmental and genetic factors – a strategy that has been adopted in lifecourse epidemiology.58 More complex analysis of lifecourse events may include investigating the effects of intervention following the occurrence of earlier epigenetic changes. A recent example of this kind of study includes that of folic acid supplementation in juvenile rats which modifies the phenotype and epigenotype first moulded by prenatal nutrition.59 Studies of this type are important as they indicate the possibility of simple nutritional intervention in modifying or reversing epigenotype within specific windows of plasticity.
Knowledge gaps
Despite rapid advances in both knowledge and technology in the field of epigenetics, there are still a number of knowledge gaps to be addressed. Some of these areas are described in table 2. In solving some of these problems, a more thorough understanding of the role of epigenetics throughout the lifecourse can be attained.
Outline of current knowledge gaps
Summary
Evidence is building to demonstrate that epigenetic mechanisms are important mediators of environmental influences on the genome. This evidence base is built largely on DNA methylation studies and there remain several fundamental issues to be addressed. The apparent epigenetic plasticity observed in early life and increasing attention paid to the developmental origins of health and disease paradigm mean that neonatal and paediatric medicine could contribute significantly to advancing knowledge in the field of epigenetics.
Acknowledgments
The authors would like to acknowledge the helpful discussion of the subject matter by Professor John Mathers.
References
Footnotes
-
Funding AG is funded by the BBSRC (BB/F007981/1) and HRE is supported by the Newcastle NIHR Biomedical Research Centre in Ageing.
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.