
Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, incurable lung disease of unknown etiology with only limited treatment options. Current paradigms of disease pathogenesis feature recurrent or prolonged epithelial injury and an ensuing inflammatory response that culminates in the appearance of activated myofibroblasts. These cells are believed central to the excessive deposition of extracellular matrix that eventually obliterates the alveolar space to cause respiratory failure. Because the factors driving the accumulation of myofibroblasts remain poorly understood, effective therapies remain elusive. This review focuses on recent understanding of myofibroblasts, including their seemingly uncontrolled proliferation and survival, their controversial origin in pathological IPF tissues, and the local biochemical and biomechanical matrix factors that drive their behavior. In addition, novel antifibrotics under development for the treatment of lung disease will be discussed. As our understanding of fibroblast and myofibroblast biology regulation expands, these cells may prove to be effective therapeutic targets.
Similar content being viewed by others


Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, incurable lung disease of unknown etiology with limited treatment options [1–3]. The diagnosis of IPF is based in part on the finding of a lung pathology consistent with usual interstitial pneumonia, which features the temporally heterogeneous juxtaposition of areas of established fibrosis and normal lung with intervening nests of fibroblasts and active matrix deposition called “fibroblastic foci” (shown in Fig. 1) [3]. Current paradigms of disease pathogenesis feature recurrent or prolonged epithelial injury and an ensuing inflammatory response that culminates in the appearance of activated myofibroblasts [4]. These cells are believed central to the excessive deposition of extracellular matrix that eventually obliterates the alveolar space to cause respiratory failure. Because the factors driving the accumulation of these cells remain poorly understood, effective therapies remain elusive. This review presents recent developments in the study of fibroblasts in IPF including the factors regulating their seemingly uncontrolled proliferation and survival, the controversial topic of their origin in pathological IPF tissues, regulation by resident lung cell populations including lymphocytes, macrophages, and epithelium, and the local biochemical and biomechanical matrix factors that drive their behavior. In addition, novel antifibrotics under development for the treatment of lung disease will be discussed. As our understanding of fibroblast and myofibroblast biology expands, these cells may prove to be effective therapeutic targets.

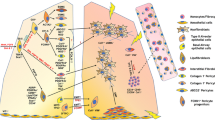
Activated myofibroblasts are believed to be central to the pathogenesis of pulmonary fibrosis. In the setting of wound repair or pathologic fibrosis, the α-SMA+ myofibroblast pool expands. Current evidence indicates that these cells are derived largely from postembryonic resident lung fibroblasts and, to a smaller extent, monocyte-derived cells called fibrocytes. The contribution of other cells such as epithelial cells (EMT), endothelial cells (Endo-MT), adipocytes (Adipo-MT) and pericytes have not been as well defined. Myofibroblast phenotypes and activity are regulated by local cells in the lung such as epithelial cells, macrophages, and lymphocytes; by intrinsic properties such as abnormalities in Caveolin-1, PTEN, AKT, FoxO3a, Activation of apoptotic and autophagic pathways; via canonical and emerging TGF-β1 dependent pathways, and by direct interactions with ECM via both biochemical and mechanosensing related pathways. The end result is the development of usual interstitial pneumonia pathology seen in the lower right corner that features the temporally heterogeneous juxtaposition of normal lung, established fibrosis, and intervening areas of active fibrogenesis called fibroblastic foci
Myofibroblasts: Role in Wound Healing and Fibrosis
Fibroblasts maintain normal tissue architecture via the regulated deposition and maintenance of the ECM components such as collagens (I, III, IV, V, VI), glycoproteins, and proteoglycans such as fibronectin, hyaluronan, and laminin [5]. They also respond to signals of tissue damage and are critical participants in tissue remodeling and wound repair. Fibroblasts display heterogeneous morphologies in tissues and are identified by their expression of ECM components. Myofibroblasts are a further differentiated subset of fibroblasts that, in addition to the above features, acquire the cytoskeletal characteristics of contractile smooth muscle cells via their production of α-smooth muscle actin (α-SMA) which distinguishes myofibroblasts from their precursor fibroblasts [6].
Current paradigms of fibrosis suggest that following exposure to endogenous or exogenous stimuli, the lung epithelium initiates an injury response resulting in the production of soluble factors such as transforming growth factor beta-1 (TGF-β1), platelet-derived growth factor (PDGF), connective tissue growth factor (CTGF), and cytokines including interleukin-4 (IL-4) and interleukin-13 (IL-13). These substances promote recruitment of inflammatory cells and mesenchymal activation which causes expansion of tissue resident post-embryonic fibroblasts which are thought to give rise to activated myofibroblasts. These cells are central to the process of wound healing but, if unmodulated, deposit excessive ECM and destroy normal lung architecture. During normal wound healing, myofibroblasts are transiently activated and direct production of granulation tissue by producing ECM and exerting traction forces. Once healing is achieved, granulation tissue is resorbed and myofibroblasts undergo programmed cell death to restore normal tissue architecture and function [5]. Disruptions at any stage in this process could cause tissue pathology. When the healing response is insufficient, as is seen in acute respiratory distress syndrome, a pathology dominated by acute injury and diffuse alveolar damage ensues. When the healing phase dominates, the tissue milieu shifts towards fibrosis and remodeling and a pathology dominated by the dysregulated accumulation of scar tissue is seen. Fibroblasts and activated myofibroblasts are believed to be central to this process [4]. This paradigm is illustrated in Fig. 1.
Pathological Fibroblast Activation in IPF
Fibroblasts and myofibroblasts in IPF demonstrate a pathologic phenotype characterized by uncontrolled proliferation and survival. These cells accumulate in lung interstitium where they deposit excessive amounts of collagen-I rich ECM and ultimately organize into the fibroblastic foci described above. As these regions expand and become juxtaposed to the alveolar space, they appear to first rupture and then ultimately destroy the alveolar basement membrane [7].
This expansion is largely attributed to the resistance to programmed cell death that has been described for primary fibroblasts obtained from IPF lung tissue [8, 9•]. Several possible mechanism(s) are proposed for this observation including abnormalities in apoptotic pathways [8, 9], aberrant Wnt signaling [10], and defective autophagy [11]. Accompanying these findings is a complementary augmentation in proliferation that in some modeling systems appears to be related to alterations in FOXO3a, an AKT inactivating transcription factor [12], abnormalities in integrin signaling [13] and dysregulated Caveolin-1 and PTEN [14]. While it has long been known that IPF fibroblasts are polyclonal and do not possess classical characteristics of malignant transformation [15], newer methods have facilitated the finding that these cells contain genome-wide abnormalities in translational control similar to changes found in cancer cells [16]. In addition to these pathways, several emerging areas of interest are the potential contribution of epigenetic alterations including DNA methylation [17] and noncoding RNAs [18, 19]. When considering the data regarding fibroblast survival and proliferation, it is important to consider these studies in light of the limitation modeling systems in which they were obtained. Similarly, it is not known whether these abnormalities are inherited or acquired or whether they contribute to fibrosis in the intact human lung. The potential contribution of these pathways is outlined in Table 1 and is demonstrated in Fig. 1.
Origin of Lung Fibroblasts
The augmented proliferation and survival described above may be accompanied by enhanced recruitment of myofibroblast precursors. In addition to tissue resident fibroblasts described above, several additional sources of lung myofibroblasts have been proposed. Of these, the best characterized sources are epithelial-to-mesenchymal cell transition (EMT), and/or recruitment and maturation of circulating pluripotent mesenchymal progenitor cells called fibrocytes [20]. A schematic representation of potential sources of IPF myofibroblasts is shown in Fig. 1.
Epithelial-to-mesenchymal cell transition, a biological process involved in cancer progression, has been proposed as a source of IPF myofibroblasts due to the invasive nature of fibroblastic foci. Early studies appeared promising, as in vitro stimulation of alveolar type II cells (AT2) with TGF-β induced expression of fibroblast markers including α-SMA [21]. Moreover, while co-localization of AT2 and fibroblast markers have been reported in IPF lung tissue these findings have been inconsistent [21, 22]. Animal studies employing cell lineage tracking of AT2 cells in the bleomycin mouse model provide little evidence to support an AT2 origin of myofibroblasts [23••]. Thus, while most sources agree that multiple stromal cell populations participate in pulmonary fibrosis, definitive evidence supporting a role for EMT (when defined by the co-expression of epithelial markers and α-SMA) remains scarce.
In contrast, robust data exist to support the notion that myofibroblasts could derive from fibrocytes, a circulating, progenitor cell population expressing the receptor for stem cell factor (CD34), monocyte-associated markers (including leukocyte-specific markers, chemokine receptors, and antigen presenting molecules) and proteins commonly produced by fibroblasts (collagens, fibronectin, and glycosaminoglycans) [24]. The potential contribution of fibrocytes to experimentally induced fibrosis and remodeling has been demonstrated in animal models [25–27] as well as in the lungs and circulation of patients with several forms of lung fibrosis including IPF [28]. However, animal modeling in the lung and other organs indicates that despite easily detectable accumulation of fibrocytes in both diseased organs and in the circulation, expression of α-SMA by these cells (which would indicate transformation into activated myofibroblasts) is scarce [20]. Thus, while a relationship between fibrocytes and lung remodeling has gained wide acceptance, their contribution to activated myofibroblasts is less defined.
As has been suggested in other organs, additional sources of lung myofibroblasts may exist including pericytes [29], adipocytes [30], bone marrow derived mesenchymal stem cells [31], and endothelial-to-mesenchymal transition [32]. To date these populations have been only minimally explored in IPF. The study of fibroblast and myofibroblast origin in the setting of fibrosis would be greatly enhanced by the generation of transgenic mice allowing lineage tracing and cell-specific deletion strategies as well as by the development of translational studies based on primary human cells.
Novel Regulators of TGF-β1
In the setting of an injurious stimulus, numerous signaling pathways are activated by which lung fibroblasts proliferate, deposit ECM, and differentiate into myofibroblasts. These signals are produced by a wide variety of cell types and many of them involve TGF-β1. TGF-β signals via the canonical SMAD-related pathways as well as an emerging array of several noncanonical, SMAD-independent mechanisms. For example, TGF-β1’s interactions with Semaphorin 7a, a neuroimmune protein expressed on a wide variety of structural and inflammatory cells, results in enhanced myofibroblast activation and fibrosis in several mouse models [33]. Interestingly, it has also been shown that Sema 7a might regulate TGF-β1 production by monocyte-derived cells in the setting of certain viral infections [34] though the relevance of this finding to myofibroblasts in the context of lung fibrosis has yet to be explored.
TGF-β1 signaling is also influenced by the local metabolic milieu of the mammalian lung. For example, one recent study found that lactic acid, the final product of glycolysis, activates TGF-β via pH-dependent mechanisms [35•]. While this study did not demonstrate a direct stimulatory effect on cultured myofibroblasts, the activation of TGF-β1 observed in this setting can be considered a surrogate for myofibroblast activation and raises the possibility that other metabolic byproducts might have similarly important effects. Last, two other recent studies find that SMAD-independent TGF-β1-induced tissue fibrotic responses are enhanced by the lectin-like carbohydrate Galectin-3 [36], and by 18-glycosyl hydrolase proteins such as Chitinase-1 [37]. When viewed in combination, these data suggest that TGF-β1-induced pathways regulated by both SMAD-dependent and novel SMAD-independent pathways work in concert to regulate myofibroblast activation and tissue remodeling responses. The potential contributions of these pathways to myofibroblast activation and fibrosis are outlined in Fig. 1 and Table 1.
Immunologic Regulation
Several types of immune responses have the potential to directly regulate myofibroblast biology as well. Innate immune agonists, such as pathogen-associated molecular patterns (PAMPs), demonstrate an increasingly important contribution to lung remodeling and fibrosis via unexpected effects on myofibroblast transformation. Toll-like receptors 2 and 4 have long been associated with enhanced myofibroblast activation and fibrosis in the setting of experimentally induced lung fibrosis [38]. More recently, high levels of TLR9 have been detected on fibroblasts obtained from the lungs of patients with rapidly progressive IPF where it appears to mediate enhanced fibroblast responsiveness to CpG islands when compared to fibroblasts obtained from patients with more stable disease [39•]. These data suggest that innate immune signaling extends beyond the initiation of inflammatory responses and point to new areas of study in the regulation of fibrotic responses.
In terms of adaptive immune responses, the potential contribution of lymphocytes is an emerging area of investigation. Animal modeling indicates that substances produced by Th1 cells, such as interferon gamma, might be sufficient to induce structural cell injury and initiate fibrotic responses. However, because Th1 cytokines exert suppressive effects on fibroblast proliferation and migration, Th1 cells would exert initiating effects on cells other than fibroblasts but would not directly influence myofibroblasts this model. Th2 and Th17 cells are also recruited to the lung following epithelial injury and they produce IL-4, IL-13, and/or IL-17 which activate STAT6 signaling within fibroblasts to induce profibrotic gene programs [2, 40, 41]. Surprisingly, evidence gleaned from several animal models suggests that regulatory T cells, which are typically thought of as antifibrotic, possess profibrotic properties under certain circumstances [42, 43•]. However, the contribution of lymphocytes to IPF remains controversial for several reasons. First, animal modeling demonstrates that lymphocytes are not required for the development of fibrosis. Second, IPF has not been typically associated with classical immune responses and is in fact defined by its lack of autoimmunity [3]. Last, treatment of IPF patients with immunomodulatory agents is associated with worsened clinical outcomes [44•]. However, these conflicting and seemingly contradictory data can be reconciled by a unifying hypothesis in which non-classical immune responses participate in IPF by amplifying existing fibrotic responses. This paradigm is shown in Fig. 1.
Macrophages also possess several functions that could influence the activity of fibroblasts including stimulation of fibroblast proliferation, recruitment, and activation, regulation and catabolism of ECM components, and secretion of cytokines and profibrotic growth factors. Macrophages may also suppress fibroblast activity including inhibitory effects on fibroblast proliferation, ECM production and transformation into activated myofibroblasts. An emerging body of evidence attributes these competing functions to classically (M1) and alternatively (M2) activated macrophages [45••, 46, 47], with M1 macrophages being antifibrotic and M2 macrophages being either profibrotic or regulatory. Indeed, our own observations indicate that scavenger-receptor expressing intrapulmonary macrophages isolated from several models of murine lung fibrosis are sufficient to induce fibroblast proliferation and survival [45••] and several recent human studies indicate that enhanced concentrations of macrophage-associated soluble mediators are associated with reduced event free survival in IPF [48, 49]. While the M1/M2 dichotomy is likely overly simplistic it is useful when framing the potential interactions between macrophages and fibroblasts. Nevertheless, while macrophages may be functionally important in the development or maintenance of IPF, many unanswered questions remain about their contributions to myofibroblast recruitment and activation. Determining their precise contribution to these processes will be challenging given the remarkable plasticity of these cells. The presumed contributions of immunologic effector cells and pathways is shown in Fig. 1.
Epithelial-Mesenchymal Interactions
Damage to the pulmonary epithelium is well accepted as the inciting event in IPF. Initial and/or repetitive injury takes many forms, including viral infections, toxic exposures including cigarette smoke, chronic microaspiration in the context of reflux, and even genetic abnormalities in surfactant proteins or telomerase [50]. Following injury, however, epithelial cells themselves produce multiple profibrotic mediators, transforming from bystanders into active participants in the fibrotic response [51]. Specifically, AT2s produce cytokines and chemokines that attract inflammatory cells and fibrocytes to areas of injury [27, 51]. They also secrete numerous growth factors including EGF, PDGF, and CTGF that promote migration, proliferation, and activation of fibroblasts [52]. They synthesize cytokines such as TNF-α that drive the initial inflammatory response as well as Th2 cytokines including IL-4 and IL-13 which activate fibroblasts and induce production of ECM components. Other products of AT2 cells might have unexpected effects on myofibroblast activation. For example, surfactant protein D, a major secretory product of AT2s, stimulates macrophage production of TGF-β1 which would be expected to stimulate myofibroblast transformation and activation [53] in the bleomycin-challenged murine lung. AT2s also directly produce TGF-β, which stimulates fibroblast activation and differentiation into myofibroblasts [52] and may participate in TGF-β1 activation via the production integrin αvβ6, which is necessary for the activation of latent TGF-β in the mesenchyme [50, 54].
In addition to their stimulatory effects on myofibroblast activation, AT2s might also participate in the suppression of myofibroblast transformation via their important role in maintaining lung homeostasis. Animal modeling indicates that the AT2 dysfunction seen in certain genetic diseases [55] or models of inducible AT2 deletion [56•] is sufficient to engender the accumulation of activated myofibroblasts and the histologic appearance of pulmonary fibrosis. This finding may be due in part to the loss of AT2 fibroblast-suppressive functions that are normally mediated by prostaglandin E2. The relationship between AT2 cells and fibroblasts is shown in Fig. 1.
Interactions with Extracellular Matrix
In addition to the interactions described above, recent evidence demonstrates that lung myofibroblasts display an important relationship with the ECM of the IPF lung. In IPF, the ECM is enriched for fibrillar Type I and III collagens, various glycoproteins, and proteoglycans [57••]. In the normal repair response, fibrillar type I collagen attenuates fibroblast proliferation via integrin-dependent mechanisms [13, 58, 59], an interaction that appears to be lacking in IPF. In terms of glycoproteins, myofibroblast differentiation in IPF may also be induced by alternatively spliced domain A containing fibronectin [60–62]. Similarly, periostin also accumulates in the IPF ECM where it may stimulate fibroblast activity [63]. In terms of proteoglycans, hyaluronic acid (HA) and HA fragments are detected at sites of tissue injury where they contribute to wound healing and fibrosis in part via the regulation of fibroblast invasiveness and ECM synthesis [64•].
While these bioactive properties are accompanied by mechanical properties that could significantly influence fibroblast phenotypes, current understanding of ECM mechanics in the intact IPF lungs is at best limited. Available ex vivo data indicate that fibroblast function and myofibroblast differentiation are enhanced by the rigidity and stiffness of the ECM [65] and that these factors might be enacted independently or in concert with classical pro-fibrotic mediators such as TGF [66, 67, 68••]. The transmission of traction forces from the cell to the ECM, and vice versa, occur through the focal adhesions (FA) connecting the cytoskeleton and ECM via transmembrane integrin receptors [69] that in the context of fibrosis signal via the Wnt/β-catenin pathway [70, 71], the Hippo pathway [72], and certain ion channels [73]. The development of modeling systems incorporating precise control of stiffness allows investigation of biomechanical properties in an in vitro setting [66, 67, 68••, 74–76]. In addition, decellularized human lung explants from IPF patients and healthy controls can be modified to create a three-dimensional culture platform retaining the native stiffness and biochemical properties of the source lung [57••]. The latter approach is attractive because it incorporates the matrix from both human diseases of interest or from experimentally induced animal models. As these models evolve to account for other physiologic aspects such as shear stress and flow dynamics, it is likely that our ability to study the interaction between fibroblasts and the lung ECM will be dramatically improved.
Targets for Therapy
Interventions at any point in the fibrotic cascade have the potential to influence myofibroblast activity and disease progression. For example, the epithelial damage responses described above might be modulated by glutathione repletion and antioxidant therapy. Indeed, as demonstrated by the IFIGENIA trial, addition of N-acetyl cysteine (NAC) to what at the time was viewed as conventional anti-fibrotic treatment with prednisone and azathioprine, led to improved outcomes [77] in subjects randomized to the treatment arm. The benefit of NAC as monotherapy is currently under investigation as part of the NIH-sponsored PANTHER trial [44•]. Another epithelial target, αvβ6 integrin, has shown benefit in animal models [78] and its utility as a therapeutic target in IPF is currently under investigation in human subjects.
In terms of inflammatory responses, interferon-gamma was tested in patients with IPF based on its ability to attenuate the profibrotic properties of fibroblasts. Despite showing promise in early phase I trials, systemic administration of IFNγ failed to show benefit in phase III trials [79]. Similarly, based on early studies suggesting a role for prednisone and azathioprine in the treatment of IPF, when this approach was tested in the recently conducted PANTHER trial, an increase in all cause mortality was seen in the subjects randomized to the treatment arm [44•] causing the data safety monitoring board to terminate this arm of the study. While it is not clear whether these outcomes were related to alterations in myofibroblast biology, these data indicate that immunomodulatory agents have the potential to exert unexpected effects on the fibrotic response and may actually indicate an as yet undefined role for inflammatory cells in the control of fibroblast responses. Current immunomodulatory strategies in clinical trials include therapies based on antibody-mediated neutralization of Th2 cytokines such as IL-4 or IL-13, as well as modulators of the M1/M2 balance such as recombinant pentraxin 5.
Strategies based on neutralization of profibrotic soluble mediators are also being investigated in IPF however the somewhat conflicting results of these studies reflect the complexity of the fibrotic response. For example, endothelin receptor signaling has proved ineffective in clinical trials despite animal and in vitro data that indicate ET production by endothelium and platelets exerts potent mitogenic and profibrotic effects on fibroblasts [80]. A phase I clinical trial is currently underway using an antibody directed against the active forms of TGF-β [81]. Given its central role in pulmonary fibrosis, neutralizing this growth factor provides an attractive potential therapy. However, because TGF-β plays important roles in modulating inflammation and autoimmunity as well as in tumor suppression, this approach may be complicated by unacceptable side effects. Similarly, the efficacy of antibody mediated anti-CTGF therapy is currently under investigation as part of a phase II in patients experiencing progression of IPF.
To date, the two most advanced agents under investigation for the treatment of IPF are pirfenidone and nintedanib. Pirfenidone, a pyridone analog, exerts potent anti-proliferative effects on cultured fibroblasts and is efficacious in preclinical studies. Several large randomized controlled trials suggested a potential benefit delaying the disease progression (reviewed in [82]). While this agent is approved for use in Europe, parts of Asia, and Canada, its approval by the FDA will depend in large part upon the results of an ongoing phase III trial in the U.S. Similarly, nintenanib is a tyrosine kinase inhibitor that in preclinical studies attenuated the activation of fibroblasts and myofibroblasts. In a recently reported phase II trial, subjects randomized to the highest dose demonstrated an impressive reduction in all-cause mortality [83]. A multicenter phase III trial testing the efficacy of this agent is currently ongoing.
In addition to these approaches, which are largely based on modulating cellular responses, interventions aimed at reducing the rigidity of healing tissues could potentially affect progression of not only IPF, but the entire class of fibrotic diseases. Several enzymes, including transglutaminases, lysyl oxidases, and prolyl hydroxylases, are known to cross-link matrix proteins thereby regulating the stiffness of fibrotic tissue. The cross-linking enzyme lysyl oxidase-like-2 (LOXL2) has been shown to be increased to a greater degree in fibrotic lung as compared to healthy tissue, and recent studies in a mouse model have shown that antibody-mediated LOXL2 inhibition decreases myofibroblast accumulation, collagen deposition, and TGF-β signaling [84]. These findings suggest that targeting the molecular pathways linking matrix mechanics to cellular functions may prove to be important therapeutic targets. Indeed, a phase I clinical trial administering inhibitory α-LOXL2 to patients with pulmonary fibrosis is currently in early stages.
Conclusion
As the ultimate effector cells in pulmonary fibrosis, activated myofibroblasts possess an array of functions that contribute to disease. Investigations focused on the regulation of proliferation, survival, activation and ECM production have the potential to heighten our understanding of IPF as do the study of regulation of these aspects by soluble mediators, inflammatory cells, and lung specific entities such as the alveolar epithelium and the ECM. A broad set of therapies based on various aspects of this response is in the pipeline. Study of these and other issues would be advanced by the development of more targeted murine modeling systems, improved access to human tissues, and the generation of bioengineering based and novel imaging techniques.
References
Recently published papers of particular importance have been highlighted as: • Of importance •• Of major importance
Murray LA, Rubinowitz A, Herzog EL (2012) Interstitial lung disease: is interstitial lung disease the same as scleroderma lung disease? Curr Opin Rheumatol 24:656–662
Homer RJ, Elias JA, Lee CG, Herzog EL (2010) Modern concepts in pulmonary fibrosis. Arch Pathol Lab Med (in press)
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA et al (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183:788–824
Homer RJ, Elias JA, Lee CG, Herzog EL (2011) Modern concepts on the role of inflammation in pulmonary fibrosis. Arch Pathol Lab Med 135:780–788
Klingberg F, Hinz B, White ES (2013) The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol 229:298–309
Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, De Wever O, Mareel M, Gabbiani G (2012) Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 180:1340–1355
White ES, Lazar MH, Thannickal VJ (2003) Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J Pathol 201:343–354
Maher TM, Evans IC, Bottoms SE, Mercer PF, Thorley AJ, Nicholson AG, Laurent GJ, Tetley TD, Chambers RC, McAnulty RJ (2010) Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 182:73–82
• Nho RS, Peterson M, Hergert P, and Henke CA (2013) FoxO3a (Forkhead Box O3a) deficiency protects idiopathic pulmonary fibrosis (IPF) fibroblasts from type I polymerized collagen matrix-induced apoptosis via Caveolin-1 (cav-1) and Fas. PLoS One 8:e61017. These authors show that the resistance to apoptosis observed in IPF fibroblasts is mediated by deficiency of FoxO3a
Chang W, Wei K, Jacobs SS, Upadhyay D, Weill D, Rosen GD (2010) SPARC suppresses apoptosis of idiopathic pulmonary fibrosis fibroblasts through constitutive activation of beta-catenin. J Biol Chem 285:8196–8206
Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, Rosas IO, Morse D (2012) Autophagy in idiopathic pulmonary fibrosis. PLoS ONE 7:e41394
Nho RS, Hergert P, Kahm J, Jessurun J, Henke C (2011) Pathological alteration of FoxO3a activity promotes idiopathic pulmonary fibrosis fibroblast proliferation on type I collagen matrix. Am J Pathol 179:2420–2430
Xia H, Seeman J, Hong J, Hergert P, Bodem V, Jessurun J, Smith K, Nho R, Kahm J, Gaillard P et al (2012) Low alpha(2)beta(1) integrin function enhances the proliferation of fibroblasts from patients with idiopathic pulmonary fibrosis by activation of the beta-catenin pathway. Am J Pathol 181:222–233
Xia H, Khalil W, Kahm J, Jessurun J, Kleidon J, Henke CA (2010) Pathologic caveolin-1 regulation of PTEN in idiopathic pulmonary fibrosis. Am J Pathol 176:2626–2637
Mio T, Nagai S, Kitaichi M, Kawatani A, Izumi T (1992) Proliferative characteristics of fibroblast lines derived from open lung biopsy specimens of patients with IPF (UIP). Chest 102:832–837
Larsson O, Diebold D, Fan D, Peterson M, Nho RS, Bitterman PB, Henke CA (2008) Fibrotic myofibroblasts manifest genome-wide derangements of translational control. PLoS ONE 3:e3220
Cisneros J, Hagood J, Checa M, Ortiz-Quintero B, Negreros M, Herrera I, Ramos C, Pardo A, Selman M (2012) Hypermethylation-mediated silencing of p14(ARF) in fibroblasts from idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 303:L295–L303
Milosevic J, Pandit K, Magister M, Rabinovich E, Ellwanger DC, Yu G, Vuga LJ, Weksler B, Benos PV, Gibson KF et al (2012) Profibrotic role of miR-154 in pulmonary fibrosis. Am J Respir Cell Mol Biol 47:879–887
Yang S, Cui H, Xie N, Icyuz M, Banerjee S, Antony VB, Abraham E, Thannickal VJ, Liu G (2013) miR-145 regulates myofibroblast differentiation and lung fibrosis. FASEB J 27(6):2382–2391
Reilkoff RA, Bucala R, Herzog EL (2011) Fibrocytes: emerging effector cells in chronic inflammation. Nat Rev Immunol 11:427–435
Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z (2005) Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol 166:1321–1332
Yamada M, Kuwano K, Maeyama T, Hamada N, Yoshimi M, Nakanishi Y, Kasper M (2008) Dual-immunohistochemistry provides little evidence for epithelial-mesenchymal transition in pulmonary fibrosis. Histochem Cell Biol 129:453–462
•• Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, and Hogan BL (2011) Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA 108:E1475–1483. This paper provides evidence that EMT is not the major source of a-SMA+ myofibroblasts in experimentally induced lung fibrosis
Peng H, Herzog EL (2012) Fibrocytes: emerging effector cells in chronic inflammation. Curr Opin Pharmacol 12:491–496
Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH (2004) Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest 113:243–252
Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB (2006) The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol 35:175–181
Garibaldi BT, D’Alessio FR, Mock JR, Files DC, Chau E, Eto Y, Drummond MB, Aggarwal NR, Sidhaye V, King LS (2013) Regulatory T cells reduce acute lung injury fibroproliferation by decreasing fibrocyte recruitment. Am J Respir Cell Mol Biol 48:35–43
Peng X, Mathai SK, Murray LA, Russell T, Reilkoff R, Chen Q, Gulati M, Elias JA, Bucala R, Gan Y et al (2011) Local apoptosis promotes collagen production by monocyte-derived cells in transforming growth factor beta1-induced lung fibrosis. Fibrogenesis Tissue Repair 4:12
Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS (2010) Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176:85–97
Uezumi A, Ito T, Morikawa D, Shimizu N, Yoneda T, Segawa M, Yamaguchi M, Ogawa R, Matev MM, Miyagoe-Suzuki Y et al (2011) Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J Cell Sci 124:3654–3664
Ueno T, Nakashima A, Doi S, Kawamoto T, Honda K, Yokoyama Y, Doi T, Higashi Y, Yorioka N, Kato Y et al. (2013) Mesenchymal stem cells ameliorate experimental peritoneal fibrosis by suppressing inflammation and inhibiting TGF-β1 signaling. Kidney Int
Hashimoto N, Phan SH, Imaizumi K, Matsuo M, Nakashima H, Kawabe T, Shimokata K, Hasegawa Y (2010) Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 43:161–172
Gan Y, Reilkoff R, Peng X, Russell T, Chen Q, Mathai SK, Homer R, Gulati M, Siner J, Elias JA et al (2011) Role of semaphorin 7a signaling in transforming growth factor beta1-induced lung fibrosis and scleroderma-related interstitial lung disease. Arthritis Rheum 63:2484–2494
Sultana H, Neelakanta G, Foellmer HG, Montgomery RR, Anderson JF, Koski RA, Medzhitov RM, Fikrig E (2012) Semaphorin 7A contributes to West Nile virus pathogenesis through TGF-beta1/Smad6 signaling. J Immunol 189:3150–3158
• Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, Honnons S, Jones C, Isern NG, Hu JZ et al. (2012) Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-beta. Am J Respir Crit Care Med 186:740–751. These authors find that metabolic by-products exert important stimulatory effects on TGFβ1 induced myofibroblast activation
Mackinnon AC, Gibbons MA, Farnworth SL, Leffler H, Nilsson UJ, Delaine T, Simpson AJ, Forbes SJ, Hirani N, Gauldie J et al (2012) Regulation of transforming growth factor-beta1-driven lung fibrosis by galectin-3. Am J Respir Crit Care Med 185:537–546
Lee CG, Herzog EL, Ahangari F, Zhou Y, Gulati M, Lee CM, Peng X, Feghali-Bostwick C, Jimenez SA, Varga J et al (2012) Chitinase 1 is a biomarker for and therapeutic target in Scleroderma-associated interstitial lung disease that augments TGF-beta1 signaling. J Immunol 189:2635–2644
Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA et al (2005) Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 11:1173–1179
• Trujillo G, Meneghin A, Flaherty KR, Sholl LM, Myers JL, Kazerooni EA, Gross BH, Oak SR, Coelho AL, Evanoff H, et al. (2010) TLR9 differentiates rapidly from slowly progressing forms of idiopathic pulmonary fibrosis. Sci Transl Med 2:57ra82. This paper provides evidence that innate immune agonists such as TLR9 stimulate myofibroblast transformation in human IPF and in mouse models of lung fibrosis
Sivakumar P, Ntolios P, Jenkins G, Laurent G (2012) Into the matrix: targeting fibroblasts in pulmonary fibrosis. Curr Opin Pulm Med 18:462–469
Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, Wynn TA (2010) Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med 207:535–552
Reilkoff RA, Peng H, Murray LA, Peng X, Russell T, Montgomery R, Feghali-Bostwick C, Shaw A, Homer RJ, Gulati M et al (2013) Semaphorin 7a+ regulatory T cells are associated with progressive idiopathic pulmonary fibrosis and are implicated in transforming growth factor-beta1-induced pulmonary fibrosis. Am J Respir Crit Care Med 187:180–188
• Lo Re S, Lecocq M, Uwambayinema F, Yakoub Y, Delos M, Demoulin JB, Lucas S, Sparwasser T, Renauld JC, Lison D, et al. (2011) Platelet-derived growth factor-producing CD4+ Foxp3+ regulatory T lymphocytes promote lung fibrosis. Am J Respir Crit Care Med 184:1270–1281. These authors show that regulatory T cells simulataneously exert pro- and anti-fibrotic effects on experimentally induced pulmonary fibrosis
• Raghu G, Anstrom KJ, King TE, Lasky JA, and Martinez FJ (2012) Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 366:1968–1977. The results of this Phase III trial show that combination therapy with prednisone, azathioprine and n-acetyl-cysteine worsens outcomes in patients with IPF
•• Murray LA, Chen Q, Kramer MS, Hesson DP, Argentieri RL, Peng X, Gulati M, Homer RJ, Russell T, van Rooijen N, et al. (2011) TGF-beta driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int J Biochem Cell Biol 43:154–162. This paper shows an important role for macrophages in the regulation of experimentally induced fibroblast activation and pulmonary fibrosis
Murray LA, Rosada R, Moreira AP, Joshi A, Kramer MS, Hesson DP, Argentieri RL, Mathai S, Gulati M, Herzog EL et al (2010) Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS ONE 5:e9683
Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, van Rooijen N, Haslett C, Howie SE, Simpson AJ (2011) Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med 184:569–581
Richards TJ, Kaminski N, Baribaud F, Flavin S, Brodmerkel C, Horowitz D, Li K, Choi J, Vuga LJ, Lindell KO et al (2012) Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 185:67–76
Prasse A, Probst C, Bargagli E, Zissel G, Toews GB, Flaherty KR, Olschewski M, Rottoli P, Muller-Quernheim J (2009) Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 179:717–723
Noble PW, Barkauskas CE, Jiang D (2012) Pulmonary fibrosis: patterns and perpetrators. J Clin Invest 122:2756–2762
Chapman HA (2011) Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu Rev Physiol 73:413–435
Selman M, Pardo A (2006) Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc 3:364–372
Aono Y, Ledford JG, Mukherjee S, Ogawa H, Nishioka Y, Sone S, Beers MF, Noble PW, Wright JR (2012) Surfactant protein-D regulates effector cell function and fibrotic lung remodeling in response to bleomycin injury. Am J Respir Crit Care Med 185:525–536
Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA et al (1999) The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96:319–328
Young LR, Gulleman PM, Bridges JP, Weaver TE, Deutsch GH, Blackwell TS, McCormack FX (2012) The alveolar epithelium determines susceptibility to lung fibrosis in Hermansky–Pudlak syndrome. Am J Respir Crit Care Med 186:1014–1024
• Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, Dave A, Engelhardt JF, Liu X, White ES, et al. (2010) Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med 181:254–263. This study uses a transgenic animal model to demonstrate that targeted deletion of AT2 cells causes pulmonary fibrosis in the otherwise normal mouse lung
•• Booth AJ, Hadley R, Cornett AM, Dreffs AA, Matthes SA, Tsui JL, Weiss K, Horowitz JC, Fiore VF, Barker TH, et al. (2012) Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med 186:866–876. This paper shows that decellularized lung scaffolds derived from fibrotic human lungs can be used as a model of fibroblast activation and phenotypes
Koyama H, Raines EW, Bornfeldt KE, Roberts JM, Ross R (1996) Fibrillar collagen inhibits arterial smooth muscle proliferation through regulation of Cdk2 inhibitors. Cell 87:1069–1078
Ivaska J, Nissinen L, Immonen N, Eriksson JE, Kahari VM, Heino J (2002) Integrin alpha 2 beta 1 promotes activation of protein phosphatase 2A and dephosphorylation of Akt and glycogen synthase kinase 3 beta. Mol Cell Biol 22:1352–1359
Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G (1998) The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol 142:873–881
Olsen AL, Sackey BK, Marcinkiewicz C, Boettiger D, Wells RG (2012) Fibronectin extra domain-A promotes hepatic stellate cell motility but not differentiation into myofibroblasts. Gastroenterology 142(928–937):e923
Kohan M, Muro AF, White ES, Berkman N (2010) EDA-containing cellular fibronectin induces fibroblast differentiation through binding to alpha4beta7 integrin receptor and MAPK/Erk 1/2-dependent signaling. FASEB J 24:4503–4512
Naik PK, Bozyk PD, Bentley JK, Popova AP, Birch CM, Wilke CA, Fry CD, White ES, Sisson TH, Tayob N et al (2012) Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 303:L1046–L1056
• Li Y, Jiang D, Liang J, Meltzer EB, Gray A, Miura R, Wogensen L, Yamaguchi Y, and Noble PW (2011) Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J Exp Med 208:1459–1471. This paper shows that ECM components importantly regulates fibroblast activation and invasiveness in murine models and primary human cells
Zhou Y, Huang X, Hecker L, Kurundkar D, Kurundkar A, Liu H, Jin TH, Desai L, Bernard K, Thannickal VJ (2013) Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest 123:1096–1108
Li Z, Dranoff JA, Chan EP, Uemura M, Sevigny J, Wells RG (2007) Transforming growth factor-beta and substrate stiffness regulate portal fibroblast activation in culture. Hepatology 46:1246–1256
Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, Tschumperlin DJ (2010) Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol 190:693–706
•• Huang X, Yang N, Fiore VF, Barker TH, Sun Y, Morris SW, Ding Q, Thannickal VJ, and Zhou Y (2012) Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol 47:340–348. These authors show that mechanotransduction importantly mediates myofibroblast differentiation
Goldmann WH (2012) Mechanotransduction and focal adhesions. Cell Biol Int 36:649–652
Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P, Munro J, Schroder E, Zhou J, Brunton VG et al (2011) Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 19:776–791
Charbonney E, Speight P, Masszi A, Nakano H, Kapus A (2011) β-catenin and Smad3 regulate the activity and stability of myocardin-related transcription factor during epithelial-myofibroblast transition. Mol Biol Cell 22:4472–4485
Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S et al (2011) Role of YAP/TAZ in mechanotransduction. Nature 474:179–183
Sukharev S, Sachs F (2012) Molecular force transduction by ion channels: diversity and unifying principles. J Cell Sci 125:3075–3083
Georges PC, Hui JJ, Gombos Z, McCormick ME, Wang AY, Uemura M, Mick R, Janmey PA, Furth EE, Wells RG (2007) Increased stiffness of the rat liver precedes matrix deposition: implications for fibrosis. Am J Physiol Gastrointest Liver Physiol 293:G1147–G1154
Wells RG (2005) The role of matrix stiffness in hepatic stellate cell activation and liver fibrosis. J Clin Gastroenterol 39:S158–S161
Tschumperlin DJ, Dai G, Maly IV, Kikuchi T, Laiho LH, McVittie AK, Haley KJ, Lilly CM, So PT, Lauffenburger DA et al (2004) Mechanotransduction through growth-factor shedding into the extracellular space. Nature 429:83–86
Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, MacNee W, Thomeer M, Wallaert B, Laurent F et al (2005) High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 353:2229–2242
Horan GS, Wood S, Ona V, Li DJ, Lukashev ME, Weinreb PH, Simon KJ, Hahm K, Allaire NE, Rinaldi NJ et al (2008) Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med 177:56–65
King TE, Albera C, Bradford WZ, Costabel U, Hormel P, Lancaster L, Noble PW, Sahn SA, Szwarcberg J, Thomeer M et al (2009) Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet 374:222–228
King TE, Brown KK, Raghu G, du Bois RM, Lynch DA, Martinez F, Valeyre D, Leconte I, Morganti A, Roux S et al (2011) BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 184:92–99
Scotton CJ, Chambers RC (2007) Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest 132:1311–1321
Gan Y, Herzog EL, Gomer RH (2011) Pirfenidone treatment of idiopathic pulmonary fibrosis. Ther Clin Risk Manag 7:39–47
Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, Brown KK, Flaherty KR, Noble PW, Raghu G et al (2011) Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med 365:1079–1087
Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M, Mikels A, Vaysberg M, Ghermazien H, Wai C et al (2010) Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med 16:1009–1017
Acknowledgments
Erica L. Herzog has received funding from HL109033 and Translational Lung Research Program at Yale School of Medicine.
Compliance with Ethics Guidelines
This article does not contain any studies with human or animal subjects performed by any of the authors.
Disclosure
Meagan W. Moore declares that she has no conflict of interest. Erica L. Herzog has served as a consultant to Boehringer Ingelheim, and received research funding from MedImmune, Sanofi, and Promedior.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moore, M.W., Herzog, E.L. Regulation and Relevance of Myofibroblast Responses in Idiopathic Pulmonary Fibrosis. Curr Pathobiol Rep 1, 199–208 (2013). https://doi.org/10.1007/s40139-013-0017-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-013-0017-8