Article Text

Abstract
Introduction Chronic lung disease, that is, bronchopulmonary dysplasia (BPD) is the most common complication in preterm infants and develops as a consequence of the misguided formation of the gas-exchange area undergoing prenatal and postnatal injury. Subsequent vascular disease and its progression into pulmonary arterial hypertension critically determines long-term outcome in the BPD infant but lacks identification of early, disease-defining changes.
Methods We link impaired bone morphogenetic protein (BMP) signalling to the earliest onset of vascular pathology in the human preterm lung and delineate the specific effects of the most prevalent prenatal and postnatal clinical risk factors for lung injury mimicking clinically relevant conditions in a multilayered animal model using wild-type and transgenic neonatal mice.
Results We demonstrate (1) the significant reduction in BMP receptor 2 (BMPR2) expression at the onset of vascular pathology in the lung of preterm infants, later mirrored by reduced plasma BMP protein levels in infants with developing BPD, (2) the rapid impairment (and persistent change) of BMPR2 signalling on postnatal exposure to hyperoxia and mechanical ventilation, aggravated by prenatal cigarette smoke in a preclinical mouse model and (3) a link to defective alveolar septation and matrix remodelling through platelet derived growth factor-receptor alpha deficiency. In a treatment approach, we partially reversed vascular pathology by BMPR2-targeted treatment with FK506 in vitro and in vivo.
Conclusion We identified impaired BMP signalling as a hallmark of early vascular disease in the injured neonatal lung while outlining its promising potential as a future biomarker or therapeutic target in this growing, high-risk patient population.
- Paediatric Lung Disaese
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known on this topic
Bone morphogenetic protein receptor 2 (BMPR2) signalling is a hallmark of pulmonary arterial hypertension.
What this study adds
Impaired BMPR2 signalling characterises vascular pathology at its earliest stage in the injured neonatal lung and can thus be considered a potential driver, treatable target and biomarker of disrupted pulmonary development in preterm infants with chronic lung disease, that is, bronchopulmonary dysplasia.
How this study might affect research, practice and/or policy
Our study demonstrates a comprehensive approach including human data and a preclinical, multilayered experimental model meticulously unravelled clinically relevant details of altered BMPR2 signalling in the developing lung undergoing prenatal and postnatal injury, thereby linking vascular pathology during alveolar septation to this critical modulator of pulmonary vessel disease.
Introduction
Chronic lung disease (CLD), that is, bronchopulmonary dysplasia (BPD) affects more than 30% of all preterm infants and is characterised by the impaired formation of the gas-exchange area as a result of prenatal and postnatal injury perturbing physiological alveolar and vascular development.1
Following the rarefication of the vascular bed that is tightly intertwined with alveolar simplification2 the development of pulmonary vascular disease (PVD) and the risk for subsequent pulmonary arterial hypertension (PAH) is one of the most severe complications in infants with BPD.3 4 Despite the undeniable clinical significance, earliest, yet disease relevant hallmarks characterising and driving the onset of PVD and later contributing to the progression into PAH, are poorly understood in the BPD context.
In adults and children, critical mechanisms leading to the development of PAH converge on bone morphogenetic protein receptor 2 (BMPR2) signalling5 6 with re-establishment of functional BMPR2 signalling representing a key therapeutic goal.7–9 Despite the compelling clinical evidence for the significance of the pathway in PVD10 11 as well as in organogenesis12 13 the role of BMPR2 in early stage vascular pathology and its relation to the most prevalent clinical risk factors that provoke chronic disease in the preterm lung (CLD) has not been comprehensively explored.14
We; therefore, investigated changes in BMPR2 expression in preterm infants with BPD by visualising RNA transcription in the diseased lung, correlating plasma proteomics in BPD infants with structural changes in lung MRI and addressing heritable traits of altered BMP signalling in BPD by the means of a genetic association study in n=1000 preterm infants. We assessed the dynamics of BMPR2 signalling as a developmentally relevant, established driver of vascular disease in response to the most prevalent prenatal and postnatal risk factors for adverse lung development using a multilayered neonatal mouse model. In this model, exposure to clinically relevant levels of mechanical ventilation (MV) and hyperoxia (O2) as pivotal postnatal risk factors15 was next combined with the prenatal impact of non-growth restricting doses of cigarette smoke (CS), known to affect one third of all pregnancies.16–18 We aimed to anchor BMPR2 signalling in the context of impaired alveolarisation by the use of platelet derived growth factor receptor alpha (PDGF-Rα) deficient mice, thereby addressing the interconnection of both pathways and the link of BMPR2 expression levels to alveolar septation and matrix formation.19 20 Finally, we investigated therapeutic strategies using FDA-approved drugs applied in PAH9 to reverse vascular pathology in the injured neonatal lung in vitro and in vivo.
Results
BMP protein expression as an indicator of early lung vascular injury in preterm infants developing BPD
Physiologically, pulmonary endothelial BMPR2 expression is increased in the developing lung of a preterm when compared with a term infant (figure 1A). In disease, we demonstrated an early decrease in BMPR2 expression in CD31 positive cells in the lung periphery of preterm infants postnatally exposed to MV in comparison to age-matched infants who died in the first hours of life (figure 1B). Partial correlation networks of log2 transformed protein expression profiles obtained from proteome analysis (day of life 28+; figure 1C) revealed a negative correlation between proteins of the BMPR2 and PDGF signalling cascade and signs of emphysema and interstitial remodelling as obtained by MRI. The network accounted for BPD, days of MV, days of O2 supplementation and GA (blue line: negative correlation, orange line: positive correlation; octagons: proteins, rectangles: clinical variables; edges significant at false discovery rates (FDR)<0.25). Assessing the impact of hereditary traits on the postnatal impairment in BMPR2 signalling, we performed genetic association analysis for 26 BMPR2-related genes in 1061 preterm infants (n=278 cases with moderate/severe BPD) considering case control status and length of oxygen and/or MV exposure. In 1016 single-nucleotide polymorphisms (SNP) out of 5632 SNPs with nominal significance (p≤0.05), classical BMPR2 polymorphisms known for their relevance in PAH were not associated with the presence of BPD or the duration of MV or oxygen exposure when accounting for multiple testing (online supplemental figure S1). One SNP (rs72917751-C), located in the SMAD7 gene in a genomic region and predicted to be relevant for increased gene transcription in several human tissues including the lung, was associated with the duration of oxygen supplementation (p=1.7×10−5, FDR=0.48; figure 1D).
Supplemental material

Decrease in lung vasculature BMPR2 expression and reduced BMP and PDGF plasma levels in preterm infants with BPD in the absence of relevant genetic variants. ISH quantification of BMPR2 and PECAM1 +double positive cells (positive cell area/total cell area; µm2/µm2) showed (A) the physiological increase in BMPR2 expression per endothelial cell in the developing preterm lung when compared with a term newborn as well as the decrease with mechanical ventilation (MV) in a preterm infant at the same age, that is, 24 weeks gestational age (GA) and (B) a significant reduction of overall pulmonary microvascular BMPR2 expression in the preterm infant undergoing postnatal lung injury induced by the exposure to MV for 2–8 weeks (24–26 weeks GA at birth; n=3/group) when compared with age-matched infants who died in the first days of life . (C) EDTA plasma samples were obtained in preterm neonates GA 25.1–30.6 weeks) with (1) and without (0) BPD at days of life 26–70 and subjected to proteome analysis (Olink proteomics). Infants were characterised for their lung structural changes at term by lung MRI (T2-weighted single-shot fast-spin-echo (ssFSE) sequences;) MRI left panel: infant without BPD, MRI right panel: infant with severe BPD in coronal plane showing interstitial enhancement and emphysematous changes. The partial correlation network of log2 transformed protein expression profile obtained by proteome analysis (day of life 28+) reveals a negative correlation between the protein levels of the BMPR2 and PDGF-Rα signalling cascade and the development of emphysema and interstitial remodelling. The network accounts for BPD, days of MV, days of O2 supplementation and GA (blue line: negative correlation, orange line: positive correlation; octagons: proteins, rectangles: clinical variables). All edges are significant at false discovery rate (FDR)<0.25. (D) Association of rs72917751-SMAD7 with time of supplemented O2. Rounded gene doses for the rs72917751 genotypes (imputed). association analysis was done using area sinus hyperbolicus transformed values of the time of supplemented oxygen (p=1.7×10−5, FDR=0.48). Association was adjusted for relatedness, gender, gestational age, birth weight <10 th percentile, and country of maternal origin. Effect size of rs72917751-SMAD7 in human genetic association analysis. Association of rs72917751 was investigated with BPD case–control status and three measures of respiratory support (area sinus hyperbolicus transformed). Phenotypes were adjusted for relatedness, gender, gestational age, birth weight <10 th percentile and country of maternal origin. Data are mean±SD **p<0.01, ****p<0.0001. BMPR2, bone morphogenetic protein receptor 2; BPD, bronchopulmonary dysplasia; ISH, RNA in situ hybridization.
Impaired BMP signalling in the developing lung in an early stage of PVD resulting from exposure to the most prevalent risk factors for lung injury in the preterm infant
In our preclinical neonatal mouse model (figure 2A), we showed that O2 and MV for only 8-hour provoke a significant reduction in pulmonary BMPR2 signalling as reflected by the three-fold reduction in lung protein levels of pSMAD 1-5-9 and its downstream target ID-1 (figure 2B). These changes occur during the earliest stage of lung vascular injury in a critical phase of alveolarisation (postnatal day (PND) 5–8), accompanied by a twofold induction of endothelial cells (EC) apoptosis and a reduction in micro vessel number (20–100 µm diameter; figure 2C). Pulmonary expression of the hypoxia inducible factor (HIF) 1α in these mice remained unchanged (online supplemental figure S2A).
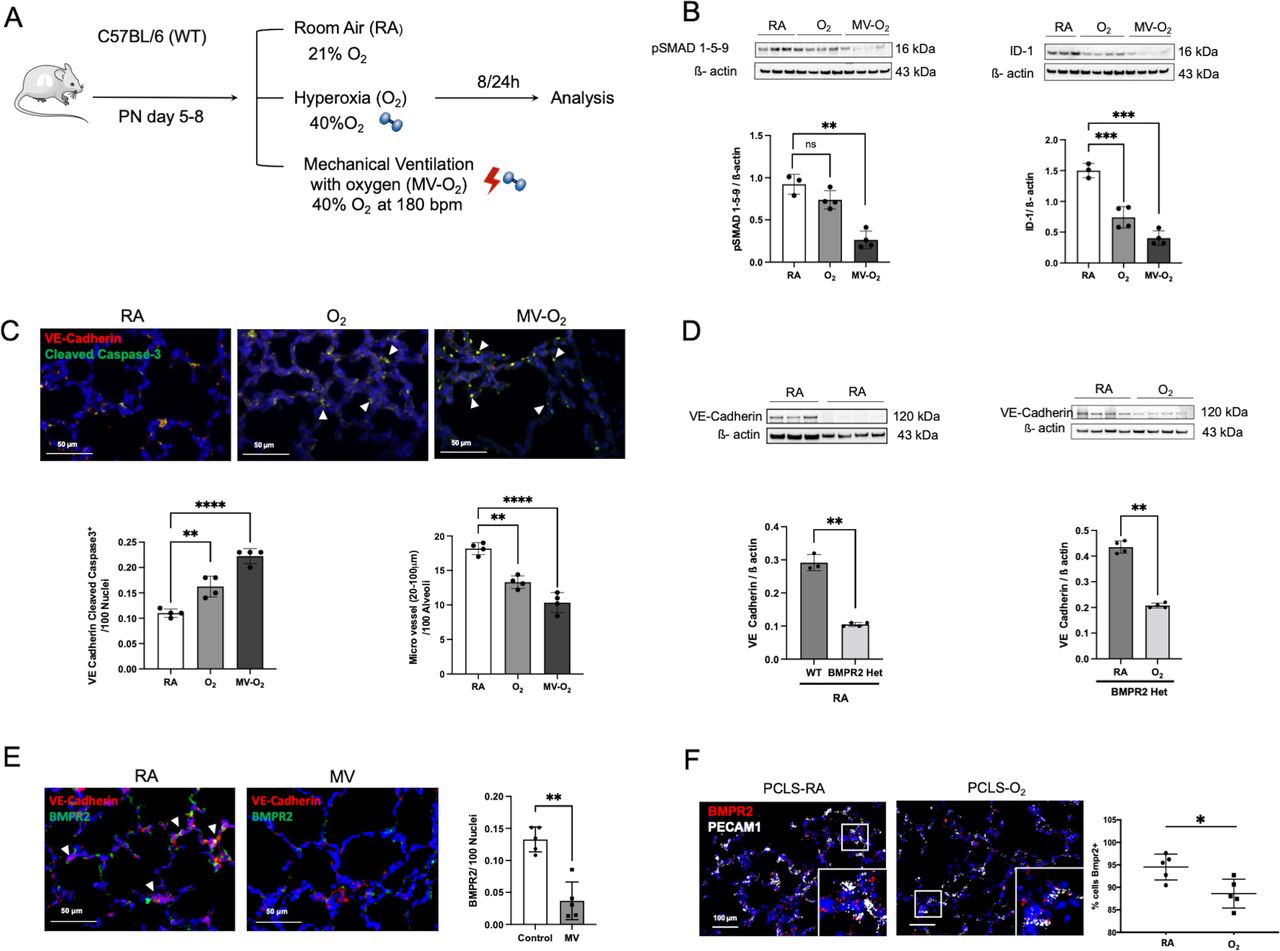
Significant reduction in BMP signalling at the earliest stage of lung vascular pathology is accompanied by microvessel loss in postnatal lung injury. (A) Preclinical mouse model of bronchopulmonary dysplasia (BPD) with induction of lung injury by exposure of neonatal mice to mechanical ventilation (MV) and/or hyperoxia (FiO2=0.4) for 8 hours or 24 hours. (B) Immunoblot analysis reveals a significant decrease in pSMAD 1-5-9 and Id-1 protein levels in WT mice in the early course of postnatal lung injury provoked by short-term MV-O2 and O2 exposure. (C) Representative images of immunofluorescence (IF) staining for cleaved caspase-3 (green) and VE-cadherin positive cells (red) in lung tissue sections (4 µM, 400X, upper panel; nuclei (DAPI, blue) from 5 to 8 day old pups show a significant increase in EC apoptosis, confirmed by quantitative analysis (lower-left panel). Histological analysis reveals a significant decrease in the number of micro vessels (20–100 µm diameter) in the lungs of mice undergoing only 8 hours of O2 and/or MV when compared with room air (RA) controls (lower-right panel). (D) In BMPR2 deficient mice, immunoblot analysis demonstrates significantly lower VE-cadherin protein expression in the neonatal lung at baseline (RA) when compared with WT pups (left panel). Postnatal exposure to moderate O2 further decreases VE-cadherin protein expression in BMPR2 deficient mice (right panel). (E) Representative images of immunofluorescence (IF) staining for BMPR2 (green) and VE-cadherin positive cells (red) in lung tissue sections (4 µM, 400X, upper panel; nuclei (DAPI, blue) following 24 hours MV show a prolonged and significant decrease in BMPR2 expression, confirmed by quantitative analysis (right panel). (F) ISH demonstrates a persistent decrease in BMPR2 transcription in neonatal mouse PCLS exposed to O2 for 24 hours when compared with RA control samples. Data are mean±SD *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, n=3–4 mice/group compared with RA controls. Quantification of if in 10 fields of view (FOV) per section in two sections per animal, normalisation of positive cells to 100 nuclei; arrows point to positive cells. ISH quantification in 212.55 µm x 212.55 µm FOV. BMP, bone morphogenetic protein; BMPR2, BMP receptor 2; ISH, RNA in situ hybridization; PCLS, precision cut lung slices; VE, vascular endothelial; WT, wild type.
A causal relationship between impaired BMPR2 and EC signalling in the neonatal lung is demonstrated by the use of BMPR2 heterozygote mice, exhibiting decreased baseline levels of lung VE-Cadherin expression and its subsequent reduction on postnatal O2 exposure (figure 2D). Indicating prolonged effects of MV and O2 on BMPR2 expression levels, we (1) confirmed the downregulation of BMPR2 expression after extended periods of MV (24 hours) by the use of IHC in vivo (figure 2E) and (2) validated these findings in vitro by imaging RNA expression levels in neonatal mouse precision cut lung slices (PCLS) showing a decrease in BMPR2 transcription in VE-Cadherin positive cells on oxygen exposure for 24 hours (figure 2F). Long-term effects of the early impairment in BMPR2 signalling are demonstrated in 18 months old animals, where lung BMPR2 signalling remained significantly decreased after only 8 hours of moderate postnatal O2 at PND 5, complying effects obtained with higher O2 levels (FiO2=0.85) (online supplemental figure S2B).
Prenatal exposure to CS resulting in increased pulmonary vulnerability to postnatal injury and aggravated impairment of BMP signalling
In order to assess the additional impact of prenatal lung injury on BMP signalling in the context of early postnatal vascular pathology, we exposed pregnant mice to non-growth-restricting doses of CS, one of the most prevalent prenatal toxins (figure 3A, online supplemental figure S3A). Proving prenatal effects of CS on the developing lung, we demonstrated increased protein levels for pulmonary cytochrome P450 1A1 (CYP1A1) and decreased levels for pulmonary secreted protein acidic and rich in cysteine (SPARC) in the lungs of these neonatal mice (PND 5–8) in comparison to filtered air (FA) controls (online supplemental figure S3B,C). Prenatal CS resulted in increased pulmonary vulnerability towards postnatal lung injury induced by O2 and MV-O2 exposure with early-stage vascular injury being tied to a significant reduction in BMP signalling, that is, decreased expression of SMAD 1-5-9 and ID1 as well as BMP4 in total lung homogenates (figure 3B). Pulmonary inflammation as a characteristic sign of lung injury was indicated by accumulation of lung monocytes/macrophages and increased TGF-β signalling following pCS exposure (figure 3C,D). These changes led to increased apoptosis (online supplemental figure S3D) with a twofold increase of VE-Cadherin and cleaved-caspase-3 double positive cells and a subsequent decrease in EC and micro vessel number (figure 3E,F; online supplemental figure S3E). Vascular pathology was further characterised by structural remodelling of the lung vessels indicated by the increased presence and adverse distribution of mature elastic fibres in the vessel wall (online supplemental figure S4A). These changes occurred in the context of alveolar simplification and were mirrored by the overall increase in smooth muscle actin (SMA) expression in the lung periphery (online supplemental figure S4B).

Early decrease in BMPR2 signalling in early lung vascular injury provoked by exposure to pCS. (A) In a preclinical BPD mouse model, neonatal mice were subjected to non-growth restricting doses of prenatal pCS followed by postnatal MV and/or O2 (FiO2=0.4). (B) Smad 1-5-9 (left panel) and ID1 (middle panel) mRNA expression show a comparable reduction after MV-O2 exposure in pCS and FA mice, whereas BMP4 is further decreased in neonatal mice exposed to pCS followed by MV-O2 as compared with FA controls (right panel). (C) Representative images of IHC stainings indicate increased presence of pulmonary monocytes/macrophages (F4/80) (4 µM, ×400, left panel) following pCS, aggravated by postnatal exposure to MV and/or O2 (right panel), and resulting (D) in a significant increase of pulmonary TGF-ß activity, that is, lung pSMAD2 protein expression. (E) Histological analysis reveals a significant decrease in microvessel number (20–100 µm diameter) induced by pCS in the lungs of neonatal mice when compared with FA controls. (F) Representative images of if staining for cleaved caspase-3 (green) and VE-cadherin (4 µM, ×400, left panel; nuclei (DAPI, blue) in lungs of 5–8 days old pups show significantly enhanced lung EC apoptosis in pCS exposed pups, aggravated byMV and O2 exposure (right panel). Data are mean±SD *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, n=3–6 mice/group. quantification of if images in 10 FOV per section in two sections per animal, normalisation of positive cells to 100 nuclei; arrows point to positive cells. ISH quantification in 212.55 µm x 212.55 µm FOV. BMPR2, bone morphogenetic protein receptor 2; BPD, bronchopulmonary dysplasia; EC, endothelial cell; FA, filtered air; FOV, fields of view; pCS, prenatal cigarette smoke.
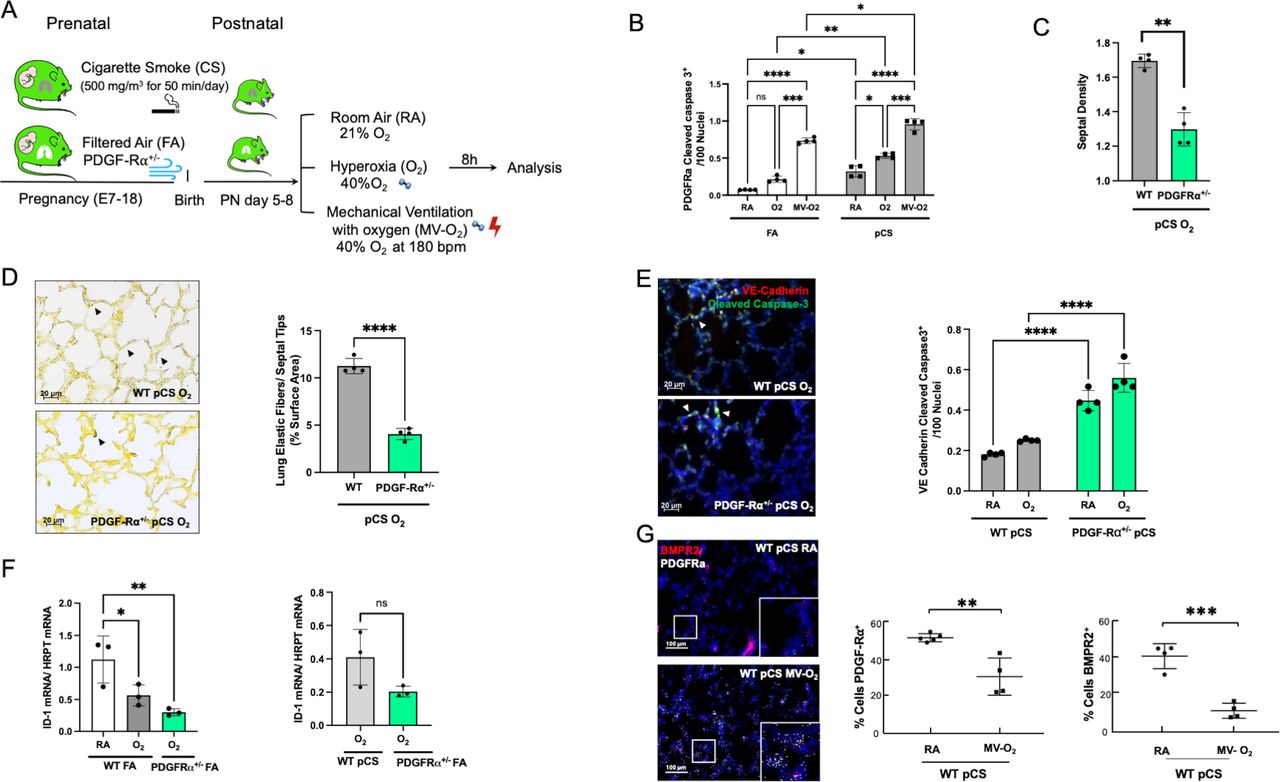
Aggravated loss of BMPR2 signalling and increased vascular pathology in the presence of BPD characteristic PDGF-Rα deficiency. (A) In our preclinical model, PDGF-Rα heterozygote (PDGF-Rα+/-) neonatal mice and WT littermates were exposed to pCS and postnatal MV and/or hyperoxia (FiO2=0.4) for 8 hours. (B) Exposure to pCS resulted in a significant increase in apoptosis of PDGF-Rα expressing cells (PDGF-Rα and cleaved caspase-3 double positive cells), aggravated by postnatal exposure to O2 or MV-O2. (C, D). The reduction in PDGFR expression is accompanied by an increased loss of septal crests and elastic fibres (Hart’s stain) on postnatal O2 exposure (4 µM, 400X) in the lungs of 5–8 days old mice. (E) Quantification of if staining in lung tissue sections show enhanced EC apoptosis in PDGF-Rα+/- neonatal mice exposed to pCS and postnatal O2, that is, increase in VE-cadherin (red) and cleaved caspase-3 (green) double positive cells (4 µM, ×400, left panel; nuclei (DAPI, blue). (F) Subsequently, mRNA analysis demonstrates decreased ID1 mRNA expression in lungs from PDGF-Rα+/- mice exposed to short-term postnatal O2, exceeding the effect in WT littermates achieved by O2 alone or in combination with pCS. (G) ISH quantification demonstrates the parallel decrease in PDGF-Rα and BMRPR2 transcription in WT mice undergoing prenatal and postnatal injury, that is, pCS and MV-O2. Data are mean±SD. *P<0.05, **p<0.01, ***p<0.001, ****p<0.0001, n=3-4 mice/group. Quantitative analysis of IF images are performed in 10 fields of view (FOV) per section in a total of 2 sections per animal, normalisation of positive cells to 100 nuclei; arrows point to positive cells. ISH quantification in 212.55 µm x 212.55 µm. BMRPR2, bone morphogenetic protein receptor 2; BPD, bronchopulmonary dysplasia; EC, endothelial cell; FA, filtered air; MV, mechanical ventilation; PCS, prenatal cigarette smoke; WT, wild type.
Impaired BMP signalling linked to PDGF-dependent alveolar pathology in the neonatal lung
Linking impaired BMPR2 signalling to the onset of defective alveolar septation, we used transgenic newborn mice to study the impact of deficient PDGF-Rα signalling on BMPR2 function in the neonatal lung undergoing prenatal and postnatal injury (figure 4A). The decrease in PDGF-Rα, a consequence of myofibroblast apoptosis during lung injury, resulted in the enhanced loss of septal quantity associated with structural abnormalities exceeding effects observed in WT littermates (figure 4B–D). These changes were accompanied by the aggravation of EC apoptosis and in the presence of impaired PDGF-Rα signalling, in line with a significant decrease in BMPR2 signalling, that is, ID1 and pSMAD 1-5-9 expression, even exceeding the significant changes provoked by preatal and postnatal environmental hazards in WT littermates (figure 4E–G; online supplemental figure S5).

Impaired BMPR2 signalling is ameliorated on treatment with FK506 in vitro and in vivo. (A) Exposure of human pulmonary microvascular endothelial cells (HPMEC) to cyclic stretch or O2 (FiO2=0.4) for 24 hours in vitro mimics postnatal injury. (B) Immunoblot analysis shows a significant downregulation of pSMAD 1-5-9 protein expression in HPMECs after exposure to O2 and/or stretch, (C) enhanced by TGF-β (D) and translated into decreased ID-1 and (E) Smad 4 protein expression. (F) Immunoblot analysis shows significant upregulation of pSMAD 1-5-9 protein levels in HPMECs on treatment with a single dose of 2 ng/mL FK506 in the presence of O2 in combination with stretch and/or TGF-β incubation. (G) The significant reduction in BMPR2 mRNA levels after O2 treatment is reversed by FK506 in HPMECs. (H) In vivo, a single intraperitoneal dose of 2 ng/mL FK506 in neonatal mice at the onset of O2 or MV-O2 exposure for 2 hours resulted in an upregulation of pulmonary CD31 mRNA expression levels in neonatal mice when compared with PBS treated control mice. Data are mean±SD *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, n=3–4 mice/group. BMPR2, bone morphogenetic protein receptor 2; ns, not significant; PBS, phosphate buffered saline.
FK506 treatment restoring impaired BMPR2 signalling in vitro and in vivo
Confirming the in vivo effects, we demonstrated EC-specific effects of O2 and stretch on human primary fetal pulmonary endothelial cells (HPMECs) in vitro (figure 5A): 24 hours exposure of HPMECs to moderate hyperoxia (FiO2=0.4), mechanical stretch or the combination of both results in a significant downregulation of pSMAD 1-5-9 expression (figure 5B). When using mechanical stretch, this effect is enhanced by TGF-β (figure 5C) with maximum effects translating into reduced ID-1 protein levels (figure 5D) together with a decrease in SMAD 4 expression (figure 5E). To evaluate treatment strategies, we subsequently show the potential of FK506 (2 ng/mL) to ameliorate or reverse the effect of stretch and/or O2 on BMPR2 signalling in HPMECs. A single dose of FK506 (2 ng/mL) results in increased pSMAD1-5-9 protein levels in HPMECs exposed to maximum injury, that is, O2 and mechanical stretch with and without additional TGF-β (figure 5F). The effect of FK506 (2 ng/mL) on BMPR2 transcription in HPMECs exposed to O2 (figure 5G) cannot be observed in human fetal fibroblasts, indicating its cell specificity (online supplemental figure S6A). A single intraperitoneal dose of 2 ng/mL FK506 increased pulmonary CD31 transcription in vivo (figure 5H; online supplemental figure S6B) and restored VEGF-R2 protein expression (online supplemental figure S6C) in newborn mice in the earliest phase of postnatal injury, that is, exposure to MV-O2 or O2 (2 hours). In summary, impaired BMPR2 signalling can be highlighted as a hallmark of early-stage vascular disease in the neonatal lung undergoing prenatal and postnatal injury, aggravated in the context of decreased PDGF-Rα signalling and persisting long term (figure 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Graphical abstract. Impaired BMPR2 signalling as a hallmark of early-stage vascular disease in the neonatal lung undergoing prenatal and postnatal injury, aggravated in the context of decreased PDGF-Rα signalling and persisting long term. We demonstrate that the exposure of the preterm lung to postnatal injury provokes an early and significant decrease in endothelial BMPR2 expression, mirrored by the joined reduction of BMP and PDGF plasma protein levels in preterm infants with a developing chronic pulmonary condition. Using multilayered preclinical models, we delineate the specific effects of the most prevalent prenatal and postnatal clinical risk factors and show early endothelial cell apoptosis and microvessel loss tied to a significant decrease in BMP signalling. The changes in BMP signalling persist long-term, are causally related to impaired PDGF-Rα expression driving septation defects and can be targeted therapeutically using an FDA approved drug. in part, the schematic was created using Servier medical art templates, creative commons Attribution V.3.0 Unported license; https://smart.servier.com. BMP, bronchopulmonary dysplasia; BMPR2; BMP receptor 2; CLD, chronic lung disease; FA, filtered air; FDA, US Food and Drug Administration; MV, mechanical ventilation; pCS, prenatal cigarette smoke.
Discussion
In this study, we demonstrated reduced pulmonary BMPR2 transcription and early postnatal reduction in related plasma protein levels in infants with characteristic lung structural changes. We addressed the impact of hereditary traits on early postnatal BMPR2 signalling in a genetic association study, and by the lack of an association suggest a role of secondary, environmental causes for the postnatal reduction in BMPR2 signalling in the preterm infant with evolving PVD.21 In subsequent experimental studies that allowed us to assess the dynamics of BMPR2 signalling in a multilayered preclinical mouse model, we successfully demonstrated the significant reduction in BMPR2 expression in early PVD caused by both prenatal and postnatal injury. We thereby established impaired BMPR2 signalling, a hallmark of PAH together with early EC apoptosis and microvessel loss, as a result of the single or combined exposure to clinically relevant environmental insults, that is, moderate levels of MV and O2 22 23 as well as non-growth restricting doses of CS.18 The mimic of clinically relevant settings significantly added to previous studies using non-physiological injury conditions.24
As demonstrated in our study, the impairment of BMPR2 signalling occurs at the earliest stage of PVD during the critical window of alveolarisation, indicating its important role in BPD-related vascular pathology as opposed to a mere end-stage phenomenon. The loss of physiologically heightened BMP receptor and ligand expression levels likely results in lasting effects,25 supported by our findings with a lack of compensatory upregulation in BMPR2 signalling after short-term injury as well as its long-term impairment in adult mice following neonatal oxygen exposure. The decrease in BMPR2 signalling can be related to BPD characteristic inflammatory processes26 in addition to the effects of oxygen25 and mechanical shear stress27 28 alone.
In line with previously published data from our group19 29 differences in response to MV/stretch in combination with O2 or O2 alone can be observed in vitro and in vivo, suggesting the activation of both incremental as well as differential mechanisms.
Successfully anchoring impaired BMPR2 signalling in the context of BPD characteristic lung structural changes, we established its interdependency with PDGF-Rα expression, that is, known to hold critical functions in alveolar septation, extracellular matrix formation and vascular development.19 20 30 31 The intertwined regulation of both pathways supports the relation of lung fibrosis and vascular pathology indicated by previous studies,32 33 aggravated by the proliferation of apoptosis-resistant ECs in the presence of impaired BMPR2 signalling34 and the subsequent exposure of subendothelial cells to growth factors and chemokines due to decreased EC barrier function.35 Uncontrolled fibroblast proliferation likely increases further in the absence of higher-functioning PDGF-Rα positive fibroblast.36 We recapitulated vascular remodelling showing the enhanced presence of mature elastic fibres in the wall of small lung vessels side-by-side with alveolar simplification and interstitial remodelling after prenatal and postnatal lung injury.19
The observed relation of PDGF-Rα and BMPR2 signalling reflects impaired pathway cross-talk as demonstrated in mesenchymal cells,37 with relevance for EC function38 and VEGF-dependent EC survival,19 39 underscoring the close relation of vascular and alveolar pathology.2 Mimicking fibroblast loss through apoptosis, reduced PDGF-Rα expression negatively affects rescue mechanisms of BMP deficiency,40 and BMP secretion by lung fibroblasts.41 Studies demonstrated the decrease of ligand-activated endocytosis of the BMPR2 receptor in the presence of VEGF deficiency, thereby arresting its signalling.42 Taken together with our findings, the lack of compensation for impaired BMPR2 signalling by, for example, HIF2 or (PDGF-Rα dependent19) VEGF-A regulation hinders the restoration of EC signalling and vascular function in the neonatal lung, including abnormal metabolic and mitochondrial cell function.25 26
The potential of BMPR2 as a therapeutic target5 6 is supported by the successful stimulation of BMPR2 signalling by the US Food and Drug Administration (FDA)-approved drug FK506 (Tacrolimus)9 in a BPD-relevant context. In line with studies in adult PAH, FK506 rescued receptor expression and enhanced EC survival in vitro and in vivo during early injury. Although previous studies were unsuccessful to prevent BPD development by nitric oxide (NO) supplementation,43 future studies in BPD should address the potency of prostacyclins, sildenafil, recombinant BMP9 or Elafin.7–9 44 45 to restore impaired BMP signalling in the BPD lung while considering the coregulation of BMPR2 signalling with endothelin, NO46 and surfactant production.47
In summary, we established impaired BMPR2 signalling in the early phase of PVD in response to the most prevalent clinical risk factors for lung injury and BPD development in preterm infants, thereby linking a hallmark of PAH with significant diagnostic and therapeutic potential to this high-risk patient cohort. The powerful combination of the most prevalent and relevant prenatal and postnatal risk factors for neonatal lung injury in a unique, multilayered preclinical animal model using wild-type and transgenic mice, enabled us to demonstrate both the dynamics as well as the clinical significance of reduced BMPR2 signalling hand-in-hand with the impaired expression of the respective signalling molecules in the circulation of the preterm infant with BPD. Aggravation of the BMP phenotype in the presence of impaired PDGF signalling points towards an important interaction of the pathways in BPD while reflecting on the interconnection of fibrosis development and vascular pathology. Testing the potential of the therapeutic stimulation of BMPR2 signalling in the injured neonatal lung, our study indicated the clinical potential of the BMP pathway as an indicator and therapeutic target of PVD in BPD.
Limitations of the study are the number of infants recruited for the translational studies, although we deem the number reasonable given the background of extreme prematurity. The lack of functional immaturity in the newborn mouse lung in comparison to the preterm infant requires additional preclinical studies to develop sufficient treatment options.
Following up on the results obtained by us, treatment strategies for preterm infant should be assessed for their potential to enhance BMP signalling, especially when aiming to improve vascular function and reduce risk for PVD. Future studies need to furthermore address the effect of reduced BMPR2 signalling in different cellular compartments of the neonatal lung, thereby providing insight into the interconnection of pathways in PVD and PAH development.
Materials and methods
Human studies
Human lung tissue analysis
Human lung tissue from autopsy specimens (EC #6 (Vanderbilt University) of preterm infants (23–26 weeks GA) that died within the first hours after birth or in the first week of life after MV and supplemental O2, respectively. Lung tissue from a term infant that died from a non-pulmonary cause served as a reference. sRNA in situ hybridisation by RNA scope (ACDBio) for BMPR2, PECAM1 (human), positive (Ppib) and negative (DapB) control was performed according to the manufacturer’s instructions. Automated analysis by Halo software (Indica Labs) from fluorescent images used Keyence BZ-X710, BZ-X Viewer software (405, 488, 561, 647 nm; 400 x). Comparative analysis was performed in two different ways: In figure 1A, each data point represents the result obtained for an individual cell quantifying the area of BMPR2 over total cell area (10 FOV/slide; 4 slides/infant). In figure 1B, each data point represents the overall %-positive BMPR2 cells in the lung periphery quantified in 10 FOV/slide, 4 slides/infant.48
Proteomic analysis in preterm plasma samples
Twenty-eight preterm infants <32 weeks GA with and without later development of BPD were included in the study (perinatal centre, LMU Hospital; EC #195-07). Plasma was derived from EDTA blood specimens at day of life 26–70. For patient characteristics, see online supplemental table S1. The infants have been characterised for lung structural changes (emphysema and interstitial remodelling) as assessed by MRI near term, that is, 36 weeks GA (3 Tesla MRI) using a 5-point Likert scale.
All samples (n=28) were randomised including inter-plate controls and measured by proximity extension assay (Olink Proteomics, Uppsala, Sweden: cardiometabolic (V.3602), cardiovascular II (V.5004), development (V.3511), inflammation (V.3012), metabolism (V.3402), neurology (V.8011), oncology II (V.7002)) following the manufacturer’s instructions followed by quantification (Fluidigm BioMark HD Real-Time PCR). NPX values were intensity normalised (plate median for each assay as normalisation factor (Intensity Normalisation V.2); samples or proteins failing quality control were excluded. Partial correlation networks were calculated using R (V4.0.0, package GeneNet (V1.2.15) with the ggm.estimate.pcor function after missing values were imputed using the imputeKNN method.49
Pulmonary MRI
Preterm infants underwent 3T MRI in spontaneous sleep at 36 weeks GA. T2-weighted single-shot fast-spin-echo sequences were quantified by a consensus panel of two senior neonatologists and two senior radiologists, blinded to the clinical diagnosis using defined criteria for scoring the presence of emphysema and interstitial enhancement on a 5-point Likert scale in four quadrants using coronal, axial and sagittal images.
Genetic association study
DNA extraction, genotyping and association analysis were performed as previously described.50 Briefly, DNA was extracted from EDTA cord blood samples of n=1061 preterm infants (n=278 moderate/severe BPD19) ≤32 weeks GA. Clinical characteristics including days of invasive and non-invasive ventilation and O2 51 were available for n=982 and 1029 infants and area sinus hyperbolicus transformed. Genotypes of patients were determined using Affymetrix Axiom microarrays (Axiom CEU array with custom content), imputation of the data used 1000 Genomes phase 1, V.3 as reference panel (genome built hg19/dbSNP 135). Association analysis of SNPs and case–control status as well as with history of respiratory support was adjusted for relatedness as previously described and accounted for gender, GA, birth weight <10 th percentile and country of maternal origin. The role of genetic variants in the BMPR2 pathway used a total of 5632 SNPs (data.frame xl5, MAF filtering) with minor allele frequency ≥1% and imputed info score ≥0.5 in or near (±5 kb) the genes ACVR1, ACVR2A, ACVR2B, ACVRL1, BMP 2, BMP 4, BMP 6, BMP 7, BMPR1A, BMPR2, CAV1, Endoglin, FKBP1A, GDF2, ID1, SMAD1, SMAD2, SMAD3, SMAD4, SMAD5, SMAD7, SMAD9, TGFB1, TGFB2, TGFBR1 and TGFBR2. Correction for multiple testing was done using FDR (Benjamini and Hochberg).
Animal studies
Animal maintenance
For breeding, pathogen-free male and female C57BL/6 (wild-type, WT) and transgenic mice were obtained from Charles River (Sulzfeld, Germany) and housed at constant temperature and humidity, a 12-hour light cycle and food and water ad libitum. All animal experiments followed strict governmental and international guidelines approved by the local government for the administrative region of Upper Bavaria or by the Stanford University Institutional Animal Care and Use Committee.
Gene-targeted mice
PDGF-Rα haploinsufficient mice (B6.129S4-Pdgfratm11(EGFP)Sor/J; no reported lung abnormalities) from Jackson laboratories (Bar Harbour, USA) were bred using heterozygous male and WT female mice to avoid effects of maternal PDGF-Rα heterozygosity. BMPR2 heterozygous mice (BMPR2+/; C57BL/6 background) were provided from Dr. Rabinovitch’s Laboratory (Stanford University). Transgenic mice were compared with WT littermates.
pCS exposure
Pregnant female mice were exposed to whole-body active 100% mainstream total particulate matter (3R4F low tar modern research cigarettes (Tobacco Research Institute, University of Kentucky, USA), 500 mg/m3, 50 min, twice/day) from day 7 to 18 of gestation (total 10 days CS) using an exposure chamber via a membrane pump.52 Controls received FA. After spontaneous delivery, pups were kept at RA with their mothers until postnatal day 5–8 (begin of experiments).
MV and O2 experiments in neonatal mice
We randomly assigned term born 5–8-day-old male and female WT and transgenic mice (WT 3.7±0.5 g; body weight (bw), PDGF-Rα+/- 3.7±0.6g; mean±SD) into three groups (13–16 mice per group), that is, 8h of MV with O2 (MV-O2, FiO2=0.4,), O2 only (FiO2=0.4) and room air controls (FiO2=0.21). As published previously.19 mice underwent (1) tracheotomy after ketamine (≈60 µg/g bw) and xylazine (≈12 µg/g bw) sedation for MV-O2 at 180 breaths/min (MicroVent 848; Harvard Apparatus, Holliston, MA; mimic of clinical conditions: mean tidal volume 8.68 µL/g bw; peak airway pressure 12-13cmH2O, mean airway pressure 11-12cmH2O) or (2) spontaneously breathed O2 or RA after sham surgery under mild sedation.19 Viable pups were euthanised (sodium pentobarbital) and lungs were harvested for analysis. For analysis of long-term effects, 5–8-day-old male and female WT mice were exposed to O2 or RA with the mothers, kept alive, weaned using a standard procedure and sacrificed at 18 months of age. Sixteen 5–8-day-old WT newborn mice (3.7 g±0.5 g bw, mean±SD) received 2 ng/mL FK506 in 100 µL/g bw 0.9% sterile saline intraperitoneally or sterile saline only before the onset of MV-O2.
Postmortem processing of lungs for quantitative histology
Lungs for histology from randomly selected mice (n=3/group) were fixed intratracheally with buffered 4%-PFA overnight @ 20 cmH2O.19 After paraffin-embedding and isotropic uniform random sectioning relative amount and distribution of insoluble lung elastin were obtained from Hart’s elastin stain as previously described.19 Representative pictures (3/animal) were obtained at 400x to assess alveolar and vascular structure.
Immunohistochemistry
Sections from 3 to 6 animals/group were incubated with peroxidase blocking reagent (Sigma, #31642), normal goat serum, primary antibodies (rat anti-F4/80; 1:400 ab6640 (Abcam)) and biotinylated goat anti-rat secondary antibody (1:200, Santa Cruz Biotechnology). Visualisation with streptavidin-HRP and diaminobenzidine (Dako, North America) was completed by counterstaining (haematoxylin (Richard-Allan Scientific, Kalamazoo, MI)) and mounting (Dako, North America, #S3023). Positively stained cells were quantified in 20 FOV/animal (400X magnification).
Immunofluorescence
Sections from (2–6 animals/group underwent antigen retrieval (0.1 M citric buffer, pH 6,≥96°C), tissue permeabilisation (Triton X-100 (0.5%), 20’) and H2O2 treatment followed by antibody incubation (4°C overnight; PDGF-Rα (C-20, Santa Cruz Biotechnology #338), VE-Cadherin (H-72, Santa Cruz Biotechnology, #28644), cleaved caspase-3 (Cell Signalling Technology, #9661). Detection with Alexa 488/568 labelled goat anti-mouse antibody (Molecular Probes; Eugene, OR; 60’) was complemented by DAPI counterstain (Sigma Aldrich #D8417). Total nuclei and single/double positive cells were quantified in 8 FOV/slide per animal (n=2–6 mice/group) (400 x; IMARIS Software, Zurich, Switzerland).
Quantitative real-time RT-PCR
Total RNA from lung tissue homogenate was isolated using the peqlab-Gold Total RNA-Kit (Peqlab, Erlangen, Germany, #12-6834-01) before cDNA synthesis (random hexamers, MuLV reverse transcriptase (Applied Biosystems, Germany)). mRNA expression of target and housekeeping gene (hypoxanthine-guanine phosphoribosyltransferase, HPRT-1) expression was determined by Platinum SYBR Green qPCR SuperMix (Applied Biosystems; StepOnePlus 96 well RTPCR System (Applied Biosystems, CA) and displayed as 2−ΔCT (ΔCT = CT target–CT reference) or as 2−ΔΔCT values (ΔΔCT = ΔCT treated–ΔCT control).
Protein extraction and immunoblot analysis
Protein was extracted from snap-frozen total lungs using high urea buffer (KPO4, Urea, AppliChem) with halt protease inhibitor (Thermo Fisher Scientific, #1861280); protein concentration measurement used BCA, Pierce Scientific Rockford, USA, #23227). Immunoblotting on Bis-Tris or Tris-Acetate gels (Life Technologies, Germany, #NP0321BOX, #EA0375BOX) included detection of CYP1A1 (A-9, Santa Cruz Biotechnology #SC-393979), ID1 (Abcam #ab168256), SMAD2/3 (Cell Signalling Technologies #3102), pSMAD2 (Cell Signalling Technologies #3101), SMAD 1 (Cell Signalling Technology, #6944), pSMAD 1-5-9 (Cell Signalling Technology, #9511), SMAD4 (Santa Cruz Biotechnology, #7966), SPARC (D10F10, Cell Signalling Technologies #8725), VE-Cadherin (H-72, Santa Cruz Biotechnology #28644), VEGF-A (147, Santa Cruz Biotechnology #507), VEGFR2 (R and D system, #AF644) and and β-actin (Santa Cruz Biotechnology, #sc-81178) (secondary antibodies goat anti-rabbit IgG (Santa Cruz Biotechnology, #2301) or goat anti-mouse IgG-HRP (Santa Cruz Biotechnology, #sc-81178)). Image quantification used chemiluminescence (ChemiDoc XRS+; GE Healthcare, Buckinghamshire, UK #RPN2232) and densitometry (Image Lab Software, Bio Rad, Munich, Germany).
In vitro experiments
Human pulmonary microvascular endothelial cells (HPMECs)
HPMECs (ScienCell Research Laboratories, CA, #3000) were cultured in ECM (ScienCell Research Laboratories, #1001) in 75 mL falcon flasks to 80%–90% confluency and seeded on flexible-bottomed laminin-coated 6 well plates (FlexCell International Corporation, #BF-3001L) for stretching experiments with 24 hours resting before experiments.
In vitro stretch
Cultured HPMEC’s underwent cyclic strain by vacuum pressure (shape/sine; elongation min 0%, max 8%; frequency 2 Hz; duty cycle 50%; cycles 43216) for 24 hours before incubation with TGF-ß (5 ng/mL) and/or FK506 (2 ng/mL).
In vitro hyperoxia
Cultured HPMEC’s underwent hyperoxia (FiO2=0.4, 5% CO2, 95% humidity, 37°C) or normoxia (FiO2=0.21) for 24 hours before incubation with TGF-ß (5 ng/mL) and/or FK506 (2 ng/mL).
Immunoblot analysis
After cell lysis with RIPA buffer and halt protease inhibitor cocktail (Thermo Fisher Scientific, #1860932), determination of protein concentration and immunoblot analysis followed the above protocol.
Precision cut lung slices
The 300 µm PCLS from 4 to 7-day-old mice were cultured in DMEM-F12 for 24 hours in O2 (FiO2=0.7) or RA (FiO2=0.21) (5% CO2, 37°C) before formalin fixation, paraffin embedding and sectioning.53
RNA scope
RNA in situ hybridisation was performed as described above using Vim, Pdgfra, BMPR2 (mouse), positive (Ppib) and negative (DapB) control (ACDBio).
Statistical analysis
Statistical analysis was performed using the Prism V.8 software package (GraphPad, San Diego, California, USA). Multiple testing used two-way analysis of variance with Bonferroni correction; comparisons of two groups/parameters used student’s unpaired t-test with Welch’s correction. Results are given as mean and SD, numbers of each experiment are presented in the figures legend).
Analysis of proteomic and genetic association data is described above in the respective method’s section.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by human lung tissue analysis: Vanderbilt University Institutional Review BoardProteomic analysis in preterm plasma samples: Ethics committees the Ludwig Maximilian-University (Ethics approval #195-07).
Acknowledgments
We sincerely thank the patients and their families of the AIRR study cohort for their significant contribution to the study by providing the samples.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @AlionderCPC
MH, PO, XZ and NK contributed equally.
Contributors Conception and design of the manuscript was done by AH and ES. The data were acquired by PO, MH, NK, XZ, EG-R, JS, AP, MK, AWF, CH, DS, XT, AOY, TP. The data analysis and interpretation were done by AH, PO, MH, KF, JS, NM, AK, TP, PA, HK and REM. The manuscript was written by MH and AH. The manuscript was drafted for important intellectual content by AH, ES, JS, TJD. AH has full access to all the data in the study and takes the responsibility for the integrity of the data and the accuracy of the data analysis.
Funding The present study was supported by the Young Investigator Grant NWG VH-NG-829 by the Helmholtz Foundation and the Helmholtz Zentrum Muenchen, Germany, the International Research Group ‘Role of BMP signalling’ (01KI07110), Helmholtz Foundation (German Ministry of Education and Health (BMBF)) and the German Centre for Lung Research (DZL, German Ministry of Education and Health (BMBF)) as well as as well as the Research Training Group Targets in Toxicology (GRK2338) of the German Science and Research Organisation (DFG) and the Progress Study Group (BMBF Grant 01KI1010C, BMBF Grant 01KI1010I).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.