Article Text

Abstract
Viral lower respiratory tract infection (LRTI) can lead to severe disease at all ages, but with the exception of influenza vaccination, prevention is not available for most respiratory viruses, hence, effective, disease-limiting therapy is urgently required. To enable the development of novel effective therapeutic approaches, we need to improve understanding of the pathological mechanisms of viral LRTI. Here, we will discuss recently gained new insight into early, innate immune and inflammatory responses to respiratory viruses by airway epithelial cells and mucosal immune cells. Following virus recognition, these cells generate a range of mediators, including innate interferons, proinflammatory cytokines, and growth and differentiation factors which have pivotal roles in effective virus control, and the development of inflammation and disease in viral LRTI.
- Viral infection
- Innate Immunity
Statistics from Altmetric.com
Viral lower respiratory tract infection (LRTI) can lead to severe disease including bronchiolitis and pneumonia, and to exacerbations of asthma and chronic obstructive pulmonary disease (COPD). With the exception of influenza vaccination, prevention is not widely available for most respiratory viruses, thus, effective therapies limiting the severity and duration of viral LRTIs are urgently required. This calls for improved understanding of the pathological mechanisms of viral LRTI. Here, we highlight recent new insights into early, innate immune and inflammatory responses to respiratory viruses (Figure 1).

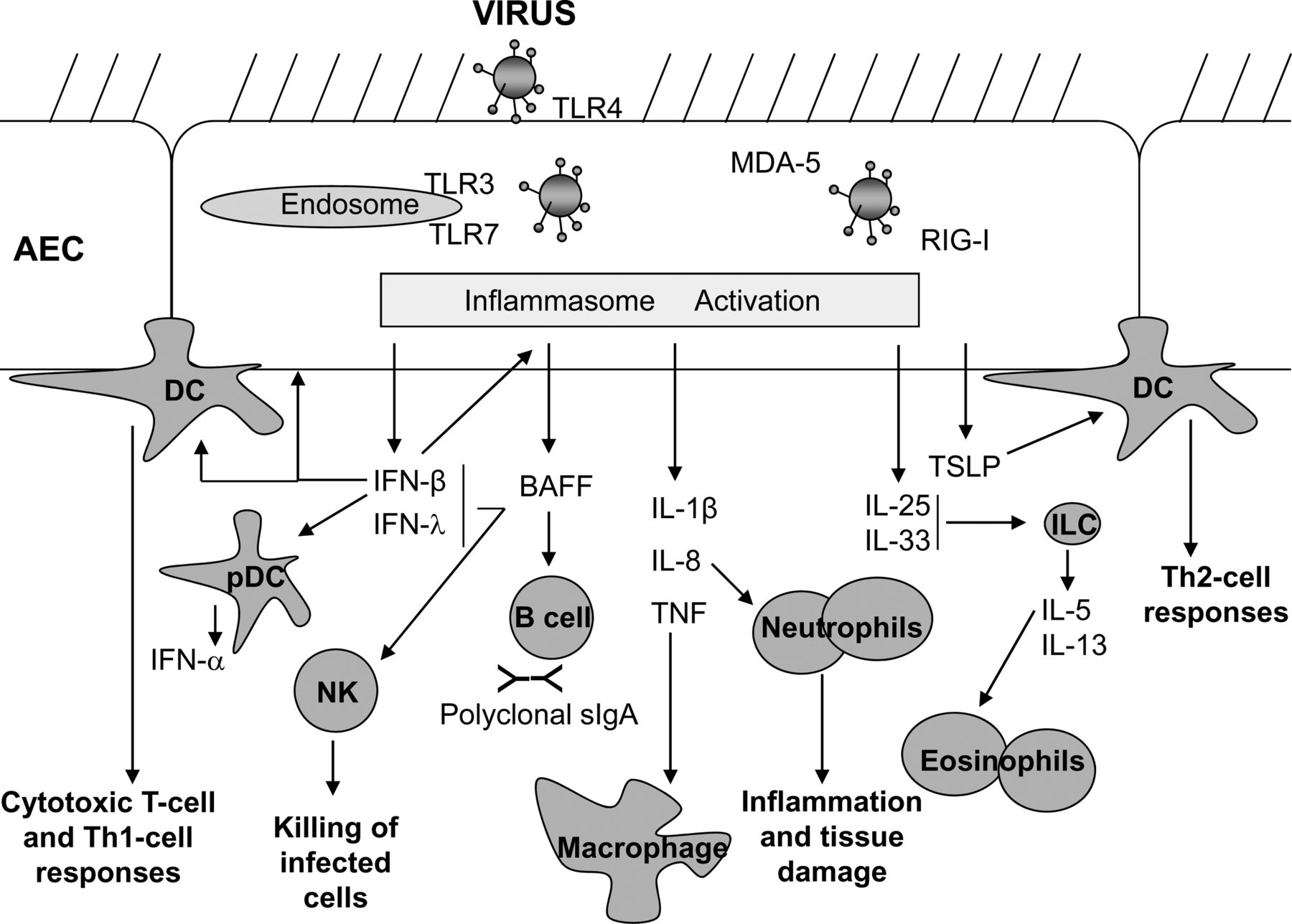{kind=link}
Respiratory viral infections can trigger a variety of pattern recognition receptors including Toll-like receptors (TLR) and RNA-helicases (e.g. RIG-I, MDA-5) on airway epithelial cells (AECs) and mucosal immune cells, resulting in the production of type-I and -III interferons (IFN). These cytokines have wide-ranging effects such as enhancing killing of virus infected cells, through programmed cell death and by activation of natural killer (NK) cells, and the production of BAFF (from AECs and dendritic cells (DCs). BAFF can promote polyclonal sIgA production which contributes to the anti-viral defence. Pro-inflammatory cytokines such as tumour necrosis factor (TNF), interleukin (IL)-I2 and IL-8 are also produced by AECs and macrophages in response to viral infection, leading to recruitment and activation of macrophages, DCs and neutrophils, all of which are involved in viral lung inflammation. The release of cytokines such as thymic stromal lymphopoietin (TSLP) from AECs primes DCs to induce Th-2 responses, and production of IL-25 and IL-33 induces release of IL-5 and IL-13 from innate lymphoid cells (ILCs), resulting in eosinophil recruitment to the lungs, goblet cell formation from AECs and mucous production and airway hyperresponsiveness. Together these innate immune responses can thus set into action a powerful inflammatory response, and will also exert a strong influence over the nature of the ensuing adaptive immune response.
Innate interferon responses
Respiratory viruses encounter airway epithelial cells (AECs) and resident mucosal immune cells, such as macrophages and dendritic cells (DCs) early after inhalation. The majority of respiratory viruses, including influenza virus, respiratory syncytial virus (RSV), rhinoviruses, parainfluenza viruses and human metapneumovirus, are RNA-viruses. Viral double-stranded RNA arising during viral replication, is recognised in human AECs by pattern recognition receptors including toll-like receptor-3 in endosomes and RNA-helicases (RIG-I, MDA-5) in the cytoplasm. This recognition leads via the NFκB transcription complex to the initial production of interferon (IFN)-β, a type-I IFN that, in an autocrine fashion enhances its own production, and initiates the production of IFN-α and (type-III) IFN-λ in AECs and innate immune cells. Together, these IFNs induce antiviral genes in AECs, trigger programmed cell death of infected cells, and activate natural killer cells, thus limiting viral replication and infection of other cells. Recent evidence shows reduced IFN responses to respiratory viral infection in AECs from patients with pre-existing inflammatory lung disease, such as asthma,1 COPD and cystic fibrosis, resulting in increased viral load and, thus, increased proinflammatory stimuli. Such patients may benefit from therapies that enhance type I/III IFN responses in viral LRTI, an effect observed in vitro using the macrolide azithromycin.2
Mucosal IgA responses
Secretory IgA antibodies (sIgA) are important in the mucosal defence against respiratory viruses. McNamara et al report in this issue of Thorax, the early, IFN-β-dependent expression of the B-cell activating factor of the TNF family (BAFF), in RSV-infected paediatric primary AECs, and its presence at high levels in bronchoalveolar lavage fluid from infants with severe RSV infection, and at lower levels in nasopharyngeal aspirates from preschool children with LRTI.3 BAFF can activate B-cells and induce their proliferation and IgA class switching. While in the absence of age-matched controls it cannot be completely excluded that BAFF levels in the airways are a function of age, these findings suggest that early BAFF production by infected AECs ensures early, non-antigen-specific B-cell activation and polyclonal sIgA production in the lung. Even upon first encounter, this would increase the chances that polyclonal sIgA may be able to bind to and neutralise a virus. Interestingly, clarithromycin, another macrolide which is used in LRTI, and which, like azithromycin, may increase type I/III IFN responses, has just been shown to enhance BAFF production from DCs, IgA class switching in B-cells, airway sIgA levels and virus neutralisation in a mouse model of influenza infection.4
Innate proinflammatory responses
Viral recognition by AECs and immune cells also results in the NFκB-dependent early production of proinflammatory cytokines, chemokines and growth factors, including IL-1β, tumour necrosis factor, IL-8, granulocyte colony stimulating factor and granulocyte macrophage colony stimulating factor. Activation of IL-1β-family cytokines requires enzymatic cleavage of a procytokine by the activated inflammasome. Triantafilou et al describe in this issue of Thorax, that the small hydrophobic envelope protein of RSV can directly activate the inflammasome, presumably by forming viral ion channels leading to changes in cellular electrolyte balance.5 Thus, RSV itself can fully activate inflammatory responses in AECs, which through their proinflammatory mediators provide important signals for the recruitment of myeloid immune cells, including neutrophils, macrophages and monocytes, and for the differentiation and activation of conventional DCs.
Myeloid immune cells
While depletion of macrophages results in enhanced disease, depletion of neutrophils prevents severe disease in a model of severe influenza (H1N1) infection.6 This suggests that, in contrast with macrophages, which may limit viral replication and inflammation, neutrophils are important drivers of tissue damage and severe inflammation, possibly through the formation of extracellular DNA nets, so-called ‘neutrophil extracellular traps’, in response to reactive oxygen species. On the other hand, early neutrophilia in influenza infection is required for sustained, effective, antiviral CD8+ T-cell responses. Interestingly, viral protein and RNA have been found in peripheral blood neutrophils in influenza and RSV infection7 raising the possibility that these cells serve as vehicles of viral replication and dissemination.
Lung DCs become activated in viral LRTI, and their numbers increase markedly for prolonged periods beyond acute infection.8 As professional antigen-presenting cells, lung DCs are thought to trigger antiviral effector and memory T-cell, and subsequent B-cell responses. However, mouse models of RSV infection suggest that they also contribute to pulmonary inflammation.9
Innate type-2 cytokine responses
It has recently been observed that viral LRTI results in the production of the innate type-2 cytokines thymic stromal lymphopoietin (TSLP), IL-25 and IL-33. TSLP can prime developing DCs to become strong inducers of Th2-cells, which can promote allergic airway inflammation and asthma. The production of TSLP and its effects on DCs are further enhanced by IL-25. IL-33, an IL-1β family cytokine, is produced by mucosal macrophages and AECs following virus-induced inflammasome activation.10 Both, IL-25 and IL-33 can activate the recently discovered innate type-2 lymphocytes (ILC-2), which are potent producers of the type-2 cytokines IL-5 and IL-13. These in turn, can lead directly, without antigen-specific T-cell responses, to eosinophilic airway inflammation, mucous hyperplasia and airway hyper-responsiveness, as has been shown in influenza infection.10 Given the close association of viral LRTI with asthma exacerbations, and in young children with an increased risk of asthma development, understanding these virus-induced innate type-2 immune responses is likely to enable the development of urgently needed preventive and disease-modifying asthma treatment.
In summary, recent research demonstrates that early responses by AECs and innate immune cells have pivotal roles in effective virus control, and in determining the severity of inflammation, tissue damage and disease in viral LRTI.
Footnotes
-
Competing interests None.
-
Provenance and peer review Commissioned; internally peer reviewed.
Linked Articles
- Respiratory infection
- Respiratory infection
- Airwaves