Article Text

Abstract
Background One hallmark of sepsis is the reduced number of lymphocytes, termed lymphopenia, that occurs from decreased lymphocyte proliferation or increased cell death contributing to immune suppression. Histone modification enzymes regulate immunity by their epigenetic and non-epigenetic functions; however, the role of these enzymes in lymphopenia remains elusive.
Methods We used molecular biological approaches to investigate the high expression and function of a chromatin modulator protein arginine N-methyltransferase 4 (PRMT4)/coactivator-associated arginine methyltransferase 1 in human samples from septic patients and cellular and animal septic models.
Results We identified that PRMT4 is elevated systemically in septic patients and experimental sepsis. Gram-negative bacteria and their derived endotoxin lipopolysaccharide (LPS) increased PRMT4 in B and T lymphocytes and THP-1 monocytes. Single-cell RNA sequencing results indicate an increase of PRMT4 gene expression in activated T lymphocytes. Augmented PRMT4 is crucial for inducing lymphocyte apoptosis but not monocyte THP-1 cells. Ectopic expression of PRMT4 protein caused substantial lymphocyte death via caspase 3-mediated cell death signalling, and knockout of PRMT4 abolished LPS-mediated lymphocyte death. PRMT4 inhibition with a small molecule compound attenuated lymphocyte death in complementary models of sepsis.
Conclusions These findings demonstrate a previously uncharacterised role of a key chromatin modulator in lymphocyte survival that may shed light on devising therapeutic modalities to lessen the severity of septic immunosuppression.
- Bacterial Infection
- ARDS
- Lymphocyte Biology
- Pneumonia
- Respiratory Infection
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Key messages
What is already known on this topic?
Lymphopenia is observed in septic patients with poor prognosis; however, our understanding on the mediators of lymphocyte death is limited.
What this study adds?
Protein arginine N-methyltransferase 4 (PRMT4) is increased in cellular and animal sepsis models and in peripheral blood leucocytes from patients with sepsis. Elevated PRMT4 protein causes caspase 3 activation and leads to lymphocyte death. Genetic depletion or chemical inhibition of PRMT4 improves mouse survival and prevents lymphocytes from death in animal sepsis models.
How this study might affect research, practice or policy?
This article reveals a novel pathway: a chromatin modulator PRMT4 as a crucial cell death mediator that causes lymphocyte death in sepsis.
Introduction
Increasing evidence has shown that an overwhelmed or hypoimmune response emerges as a critical factor contributing to the poor prognosis of patients with sepsis.1 2 Immunosuppression can result from ‘immune paralysis’ and is one sepsis hallmark that is increasingly appreciated and can also be observed in chronic infection and cancer.3 4 Typical features of immunosuppression include a reduced population of immune cells, exhaustion and dysfunction of cell immunity, suppression of proinflammatory factor release or an increase in anti-inflammatory factor release.5 6 Immunosuppression is clinically manifested in patients by chronic or recurrent bacterial and viral infections, or acute infections with opportunistic pathogens leading to sepsis. Lymphopenia has emerged as a prominent feature in septic patients that is associated with a poor prognosis. Lymphocyte death has been observed in Gram-negative bacteria derived lipopolysaccharide (LPS)-induced injury in cellular or acute animal models.7–10 Yet, the underlying molecular mechanism of lymphopenia in sepsis remains to be defined.
Epigenetics govern DNA accessibility and concomitantly coordinates with transcription factors to control lymphocyte development, lineage differentiation and maturation.11 Epigenetic alterations occur in multiorgan failure, animal models of sepsis and in critically ill patients.12–15 Bacterial infection regulates the behaviour of epigenetic enzymes to reprogramme host defences in inflammatory gene transcription, cell death and survival.16 Histone H3K27 methylation has been previously implicated as an epigenetic mark in septic immunosuppression.17 Epigenetic regulation is crucial to the pathogenesis of immunosuppression, and persistent epigenetic changes have been reported in exhausted T cells in chronic viral infection animal models.18 Nevertheless, our understanding of the role of individual epigenetic enzymes in sepsis is limited as mechanisms are poorly described.
In this study, we identified that microbial factors (ie, endotoxin) increase a type I protein arginine methyltransferase protein arginine N-methyltransferase 4 (PRMT4) expression, thereby inducing lymphocyte death, and modulate host survival in experimental sepsis. The epigenetic enzyme PRMT4 regulates crucial life processes including gene transcription, proliferation, RNA splicing and development.19–23 Knockout (KO) of PRMT4 in mice leads to neonatal death and developmental defects in the respiratory system, reduced percentage of CD4–CD8 double-negative T cells and a block in thymocyte development in mice.24–26 Our data show that bacterial pathogens increase PRMT4 in both B and T lymphocytes and monocytes. Further, we observe that PRMT4 triggers lymphocyte death, and by screening putative small molecule inhibitors of PRMT4, we identified that one compound, TP064, specifically attenuates lymphocyte cell death and protects mice after LPS lung injury and in polymicrobial sepsis.
Methods
Cell lines and reagents
Human lymphoma Jurkat cells, SKW6.4 cells, THP-1 monocytes were purchased from American Type Culture Collection (ATCC). These cells and human primary pan-T cells (StemCell) were cultured with RPMI1640 (Gibco) containing 10% FBS. Escherichia coli was from ATCC. PRMT4 (cat# 12495), cleaved caspase 3 (cat# 9661) and cleaved caspase 9 (cat# 7237) antibodies were from Cell Signalling (Danvers, Massachusetts, USA). The lenti-PRMT4 shRNA was from Origene (Rockville, Maryland, USA). β-actin (cat# A3853) antibody, bacterial LPS) from E. coli O111:B4 (cat# L4391, lot 115M4090V) were from Sigma (Carlsbad, California, USA). TP064 (cat# 6008) was from Tocris Bioscience (Ellisville, Missouri, USA). All other reagents were of the highest grade available commercially.
Cloning and plasmid transfection
PRMT4 were cloned into pcDNA3.1D-His-V5-TOPO plasmid using PCR-based approaches as previously described.16 The accuracy of the insert was confirmed by DNA sequencing. The PRMT4 primers used in plasmid construction were forward primer 5′-CACCATGGCAGCGGCGGCAGCG-3′ and reverse primer 5′-CATCAGGATCCGGATGTCAAATG-3′. Plasmids were introduced into cells using electroporation executed with a nuclear transfection apparatus (Amaxa Biosystems, Gaithersburg, Maryland, USA) in a preset program (X-001 for Jurkat cells), following the manufacturer’s instructions as previously described.27
Immunoblotting
Immunoblotting was conducted as previously described.27 Briefly, for immunoblotting, whole cell extracts (normalised to total protein concentration) were resolved by sodium dodecyl sulfate-polyacrylamide electrophoresis (SDS-PAGE) and transferred to nitride cellulose membranes by electroblotting. The membranes were blocked with 5% (w/v) non-fat milk in Tris-buffered saline and probed with a primary antibody as indicated (PRMT4 at 1:1000 dilution for 2-hour incubation) and a secondary antibody at 1:5000 dilution. Membranes were developed by an enhanced chemiluminescence system, and images were acquired using a Bio-Rad ChemiDoc XRS+ system.
Human study and PRMT4 ELISA assays
Subjects with or without sepsis were selected from a cohort of mechanically ventilated patients at the University of Pittsburgh Medical Center. Eligible patients were 18 years or older with acute respiratory failure requiring mechanical ventilation via endotracheal intubation with sepsis as defined by the presence or suspicion of infection and two or more systemic inflammatory response syndrome criteria. Control patients were intubated and mechanically ventilated for airway protection without sepsis. Deidentified baseline clinical data are presented in table 1. Deidentified plasmas from the aforementioned samples were used for assay of PRMT4 expression using PRMT4 ELISA kit (cat# MBS3244080; MyBioSource, South Carolina, USA) as directed by the manufacturer.
Patient characteristics
Single-cell RNA sequencing (scRNA-seq)
Mouse (strain C57BL/6J) CD4+ T cells were enriched by magnetic beads then activated with plate-bound anti-CD3/CD28 overnight. Activated T cells were mixed with naïve unstimulated T cells and scRNA-seq library was prepared by using the 10× Genomics Chromium Single Cell 3′ Reagent kits, sequenced on an Illumina Novaseq (Illumina, California, USA), and data were processed with Cell Ranger V.5.0 then analysed using Seurat R package. Gene expression was shown as two-dimensional uniform manifold approximation and projection plots.
Mouse splenic lymphocyte isolation, flow cytometry and annexin V apoptosis analysis
Mouse (strain C57BL/6J) spleens were disrupted in PBS containing 2% fetal bovine serum (FBS). Splenic lymphocytes were isolated using EasySep T cell or B cell Isolation Kits (cat# P19851, cat# 19854; Stemcell Technologies, Vancouver, Canada). The aforementioned isolated lymphocytes were stained with anti-CD4, CD8a or CD45R antibodies combined and mixed with annexin V binding buffer. The samples were acquired on the LSRII flow cytometer (BD Biosciences, Michigan, USA), and the data were analysed with FlowJo software (Tree Star, Oregon, USA). FITC Annexin V Apoptosis Detection Kit with propidium iodide (cat# 640914; BioLegend, California, USA) was employed to detect apoptotic cells in Jurkat cells. The samples were analysed through BD Accuri C6 flow cytometer (BD Biosciences).
Mouse LPS-induced lung injury and cecal ligation and puncture (CLP) procedures
LPS- induced lung injury model was conducted as previously described.26 C57BL/6 J mice at the age of 10 weeks were used for the experiments. LPS (7 mg/kg) were intratracheally administrated and the mice were observed for 48 hours. Polymicrobial sepsis was induced in mice by CLP. Briefly, mice were anaesthetised throughout the experiment. A midline abdominal incision of 1–2 cm was performed. The cecum was exposed and ligated with a sterile silk suture 1 cm from the tip and double punctured with a 19-gauge needle. The cecum was gently squeezed to extrude a small amount of faecal material and was returned to the peritoneal cavity. The incision was closed with silk sutures. Mice were resuscitated with intraperitoneal injection of 1 mL of prewarmed 0.9% saline solution. Mice were then monitored every 12 hours for survival or euthanised at different time points for analysis of different parameters.
Lentivirus production
Lentiviruses were generated by Lenti-X Packaging Single Shots VSV-G (cat# 631276; Clontech, California, USA) according to the manufacturer’s procedures. Lentivirus-containing supernatants were concentrated using Lenti-X Concentrator (cat# 631231). Concentrated lentiviruses were resuspended in PBS and titrated by Lenti-X GoStix (cat# 631280). The samples were aliquoted and stored at −80°C.
CRISPR/Cas9 PRMT4 KO cell line
Jurkat cells were cotransfected with PRMT4 CRISPR/Cas9 KO plasmid (cat# sc-404087-KO-2) and HDR plasmid (cat# sc-404087-HDR-2) using UltraCruz Transfection Reagent (cat# sc-395739). Stable KO cells were selected by puromycin following instructions from the CRSPR/Cas9 manufacturer. PRMT4 KO was confirmed by immunoblotting.
Statistics
Data represent the mean±SD in the graphs depicting the error bars or as specifically indicated. Prism V.7 (GraphPad Software, San Diego, California, USA) was used to determine statistical significance. Comparisons between groups were made using unpaired, two-tailed Student’s t-test and one-way analysis of variance (ANOVA). Multiple comparisons are conducted using two-way ANOVA. For animal survival experiments, a sample size of 16 in each group has 80% power to detect an HR of at least 3 (ie, overall survival of 0.81 in LPS+TP064 vs 0.40 in LPS+PRMT4 group). This sample size has >90% power to detect an overall survival of 0.94 in the LPS+shPRMT4 group vs 0.56 in the LPS group (type I error=0.05, two-sided test) (PASS 15, NCSS, LLC). Kaplan-Meier estimates were used for survival analysis in mouse septic models. Meta-analysis among murine replication experiments was conducted with random-effects restricted maximum likelihood (REML) model. Another approach was to combine two studies in one and adjust for the study effect (Stata). P values of less than 0.05 were considered as significant.
Results
PRMT4 protein expression increases in human septic patients and in septic models
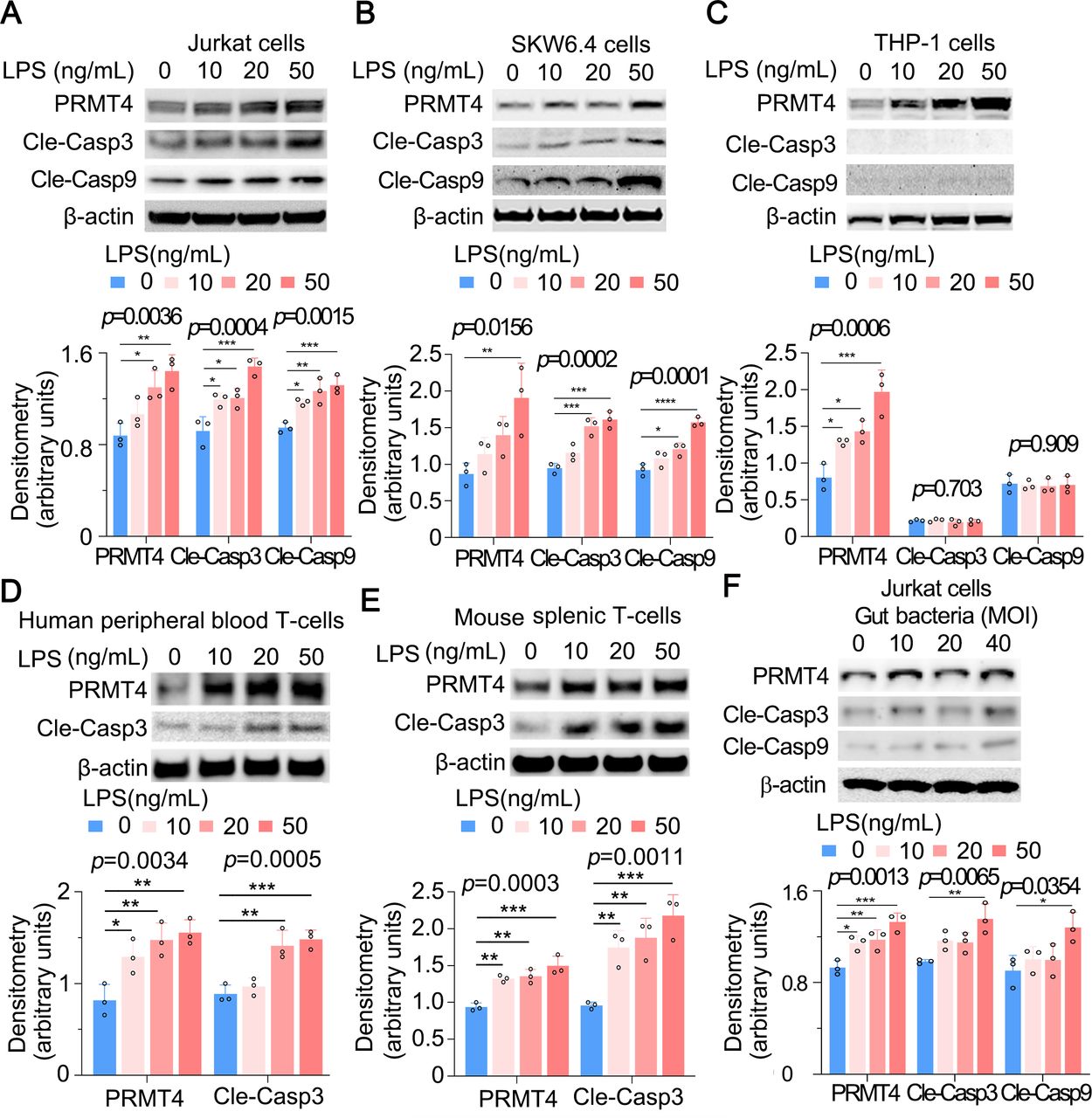
T lymphocytes have been previously reported to respond to LPS, but the functional sequelae following LPS stimulation is not fully understood.28 29 To understand whether LPS regulates PRMT4 in T lymphocytes, we first identified that E. coli-derived LPS increased PRMT4 protein levels in human lymphoma Jurkat T cells (figure 1A), human lymphoma SKW6.4 B cells (figure 1B) and monocyte THP-1 cells (figure 1C). LPS stimulation increased PRMT4 expression in a concentration-dependent manner in these cells. In addition, live E. coli increased PRMT4 cellular concentrations in Jurkat cells (figure 1D). We analysed PRMT4 in human peripheral blood leucocytes, in which approximately one-third of cells are lymphocytes and monocytes and observed a tendency in higher PRMT4 protein expression compared with that of non-septic diseased controls (figure 1E). However, PRMT4 protein levels were substantially higher in the plasma from septic patients as compared with those without sepsis (figure 1F). PRMT4 protein levels were remarkably higher in sera from polymicrobial infected mice as compared with those of untreated control mice (figure 1G). In LPS treated mice, PRMT4 was expressed in bronchoalveolar lavage fluid agranular cells but less in granular cells (PRMT4 positive cells: 96.0% in agranular cells vs 7.7% in granular cells) (figure 1H,I). In addition, PRMT4 expression was comparable in CD4+ and CD8+ subpopulations (figure 1J). Hence, these data suggest that PRMT4 expression is induced in human peripheral blood leucocytes with sepsis, and PRMT4 levels are increased during murine sepsis in vivo.
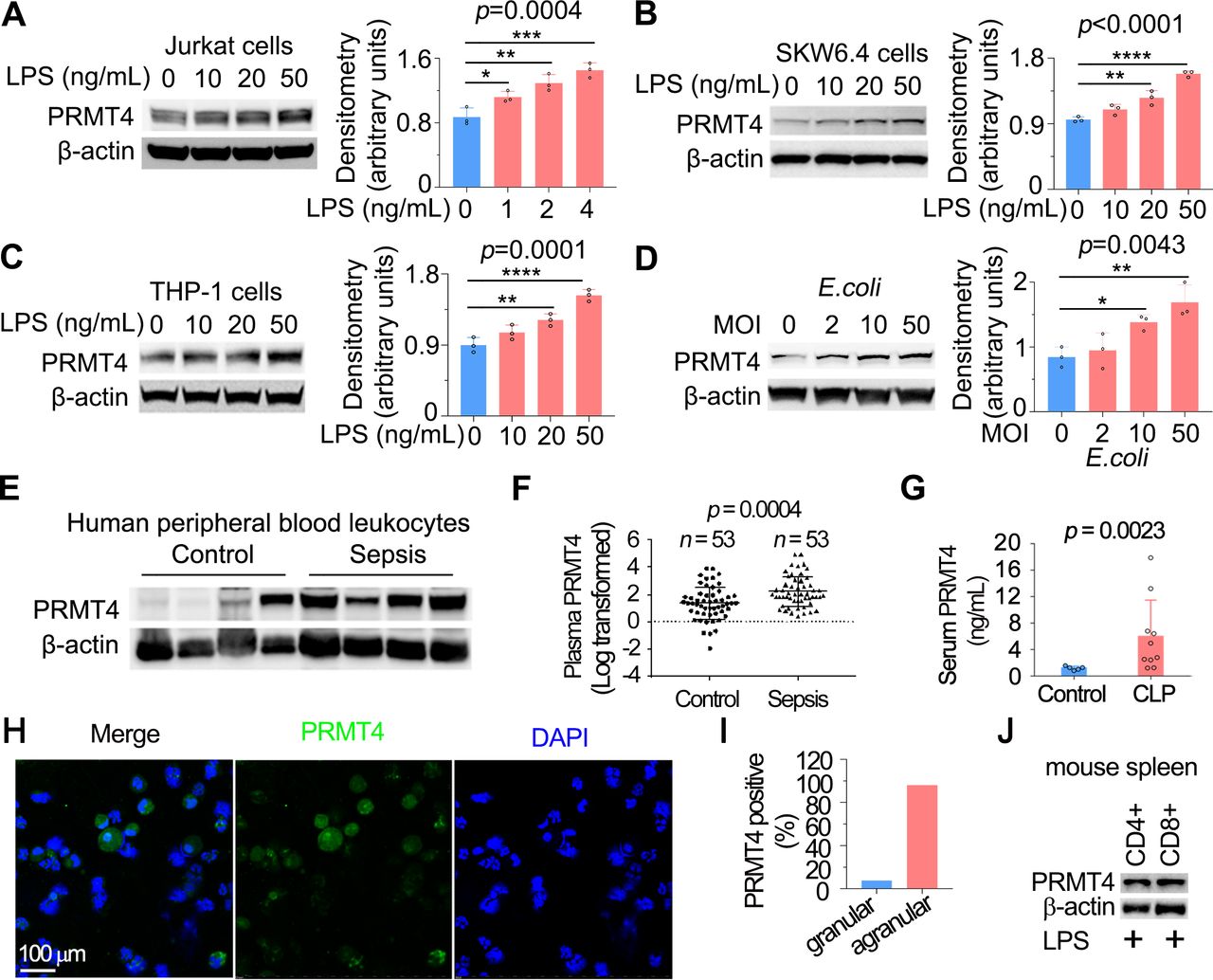
LPS and Escherichia coli increases PRMT4 protein expression in lymphocytes in vitro, and PRMT4 is increased in experimental septic models. (A–C) Jurkat cells (A), SKW6.4 cells (B) and THP-1 cells (C) were treated with LPS as indicated, and cell lysates were immunoblotted with PRMT4 and β-actin antibodies. Independent experiments, n=3. (D) Jurkat cells were treated with live E. coli as indicated, and cell lysates were immunoblotted with PRMT4 and β-actin antibodies. Densitometry was plotted in the lower panel. Independent experiments, n=3. (E) Lysates of peripheral blood leucocytes from deidentified human samples with or without sepsis were immunoblotting analysed with PRMT4 and β-actin. (F) PRMT4 protein levels were determined by ELISA from blood plasma from septic patients (n=53) and non-septic control patients (n=53). Lines indicate the median and IQR, Mann-Whitney U test, p=0.0004. (G) CLP procedures were subjected to C57BL/6 J mice for 48 hours; mice sera were collected from untreated controls (n=5) and polymicrobial infected mice (n=10) for PRMT4 ELISA analysis. (H,I) Leucocytes isolated from BALF in LPS-treated mouse were immunofluorescent stained with PRMT4 antibody. PRMT4 expression was visualised using confocal microscopy; the nuclei were stained by DAPI (H). Total cells were counted and positively stained granular and agranular cells were presented as percentage (I). A total of 300 granulocytes and 100 agranulocytes were counted. (J) Isolated CD4+ and CD8+ cells from LPS-treated mouse were lysed and immunoblotting analysed with PRMT4 antibody. Independent experiments, n=3. Scale bar=100 µm. *P=0.05–0.01, **P=0.01–0.001, ***P=0.001–0.0001, ****P<0.0001. BALF, bronchoalveolar lavage fluid; CLP, cecal ligation and puncture; DAPI, (4′,6-diamidino-2-phenylindole); LPS, lipopolysaccharide; PRMT4, protein arginine N-methyltransferase 4.
PRMT4 gene expression increases on activation in CD4+ cells
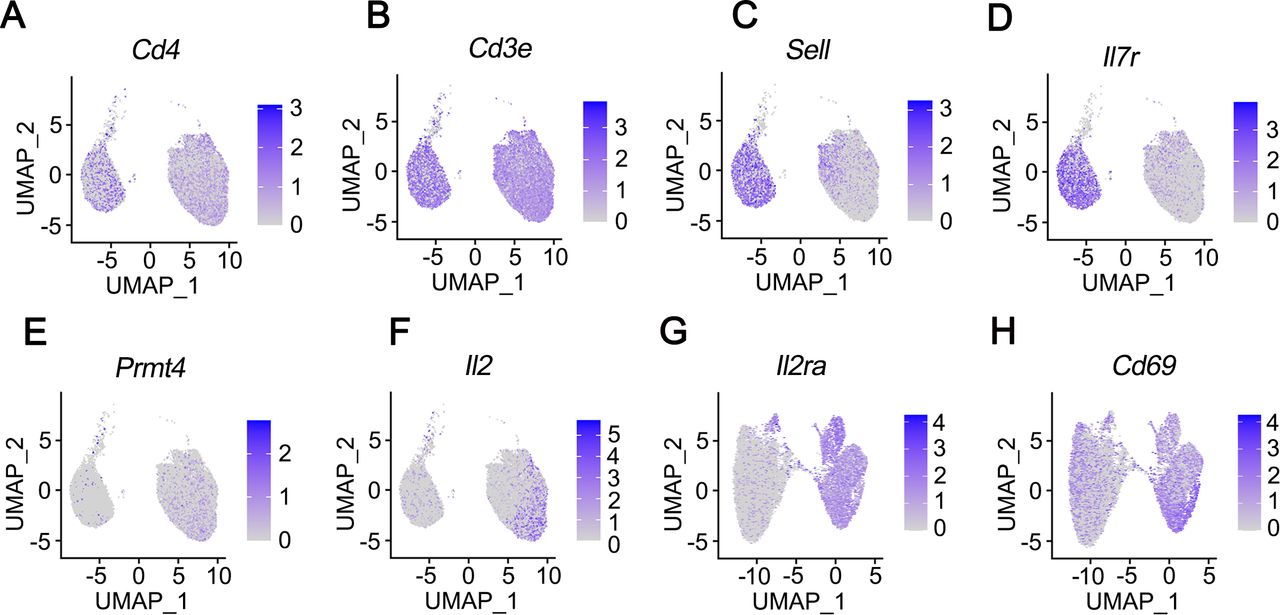
PRMT4 gene is expressed in B cells and monocytes and less so in naïve T cells (The Human Protein Atlas, https://www.proteinatlas.org/ENSG00000142453-CARM1/celltype). However, PRMT4 is necessary for T-cell lineage development as reported in PRMT4−/− mice.25 To address if pathogen may induce PRMT4 gene expression in T cells, we conducted scRNA-seq in CD4+ T cells with or without ligand activation. Mouse splenic CD4+ cells were activated with anti-CD3/CD28 overnight (figure 2). Naïve T-cell markers Sell and IL7r decreased in activated T cells, suggesting that T-cell activation via CD3/CD28 was successful (figure 2C,D). Anti-CD3/CD28 activation increased PRMT4 gene expression in T cells (figure 2E). The expression of IL2, IL2ra and CD69 was increased in activated T cells as well (figure 2F–H). These observations indicate that PRMT4 is expressed in activated T cells.

PRMT4 gene expression increases on activation in CD4+ T cells. CD4+ cells were isolated from the spleen of a mouse (strain C57BL/6J). The mixture of naïve, unstimulated T cells and CD4 T cells activated with anti-CD3/CD28 comprising a total of 10 000 cells each were applied to single-cell RNA sequencing. UMAP lots as two dimensional were used to plot the expression of CD4-specific genes CD4 (A) and CD3e (B), naïve T cell-specific genes Sell (C) and IL7r (D), CD4+ cell activation increased PRMT4 (E), IL2 (F), IL2ra (G), as well as CD69 (H) gene expression. PRMT4, protein arginine N-methyltransferase 4; UMAP, uniform manifold approximation and projection.
LPS-induced PRMT4 promotes caspase 3 activation in lymphocytes
We next assessed a potential pathophysiological role for elevated PRMT4 after bacterial infection. PRMT4 triggers apoptosis of human retinal pigment epithelial cells via H3R17 dimethylation in high-glucose treatment.30 In addition, LPS induces lymphocyte death in cellular and acute animal models.7–10 Thus, we tested if actions of LPS on lymphocyte death are mediated by PRMT4. As predicted, both caspase 3 and caspase 9 were activated in Jurkat cells and SKW6.4 cells, and the activation occurred in an LPS concentration-dependent manner, suggesting that PRMT4-mediated Jurkat cell death is possibly via the intrinsic apoptotic pathway (figure 3A,B, middle panels). Notably, LPS increased PRMT4 protein expression but without detection of cleaved caspase 3 and caspase 9 in THP-1 monocytes, suggesting a different mechanism whereby LPS stabilises PRMT4 in cells with distinct outcomes between lymphocytes and monocytes (figure 3C). We observed similar results that LPS mediates caspase 3 activation in human peripheral blood pan-T cells and mouse splenic T cells (figure 3D,E). We then treated Jurkat cells with gut-derived live bacteria (bacteria overnight cultured in an Luria-Bertani (LB) plate from the faecal material of mouse cecum) and observed that PRMT4, cleaved caspase 3 and cleaved caspase 9 are upregulated (figure 3F). These data suggest that LPS increases PRMT4 in lymphocytes and monocytes, but caspase 3 activation appears to be cell-type specific.

LPS increases PRMT4 expression and activates caspase 3 in lymphocytes. (A–C) Jurkat cells (A), SKW6.4 cells (B) and THP-1 cells (C) were treated with LPS as indicated. Cell lysates were subjected to immunoblotting for PRMT4, cleaved caspase 3, cleaved caspase 9 and β-actin. The densitometric results were plotted in the lower panels. Independent experiments, n=3. (D,E) Primary mouse splenic lymphocytes (D) and human peripheral blood T cells (E) were treated with LPS as indicated. Cell lysates were analysed by PRMT4, cleaved caspase 3 and β-actin immunoblotting. The plotted data are shown in the lower panels. Independent experiments, n=3. (F) The faecal material from mouse cecum was cultured in an LB plate overnight. Jurkat cells were treated with aforementioned gut-derived live bacteria for 2 hours. Cell lysates were immunoblotting analysed with PRMT4, cleaved caspase 3, cleaved caspase 9 and β-actin. The plotted data are shown in the lower panel. Independent experiments, n=3. *p=0.05–0.01, **p=0.01–0.001, ***p=0.001–0.0002, **** p=0.0001. LPS, lipopolysaccharide; PRMT4, protein arginine N-methyltransferase 4.
We then observed that PRMT4 ectopic expression was sufficient to activate caspase 3 in Jurkat cells (figure 4A). Caspase 3 activation occurred in a PRMT4 plasmid concentration-dependent manner. We observed similar results in SKW6.4 cells (figure 4B) but not in THP-1 monocytes (figure 4C). Furthermore, ectopic expression of PRMT4 promoted LPS-induced caspase 3 activation (figure 4D). Next, we generated PRMT4 KO Jurkat cells using CRISPR/Cas9 to test our observations (figure 4E). KO of PRMT4 blocked LPS-mediated caspase 3 activation to baseline levels (figure 4F). Results from the isolated mouse splenic T cells showed that expression of PRMT4 promotes LPS-induced caspase 3 activation, and depletion of PRMT4 by shRNA substantially blocked LPS-induced caspase 3 activation (figure 4G). Thus, these data suggest that LPS increases PRMT4 expression, thereby facilitating the methyltransferase to trigger caspase 3 signalling in lymphocytes.
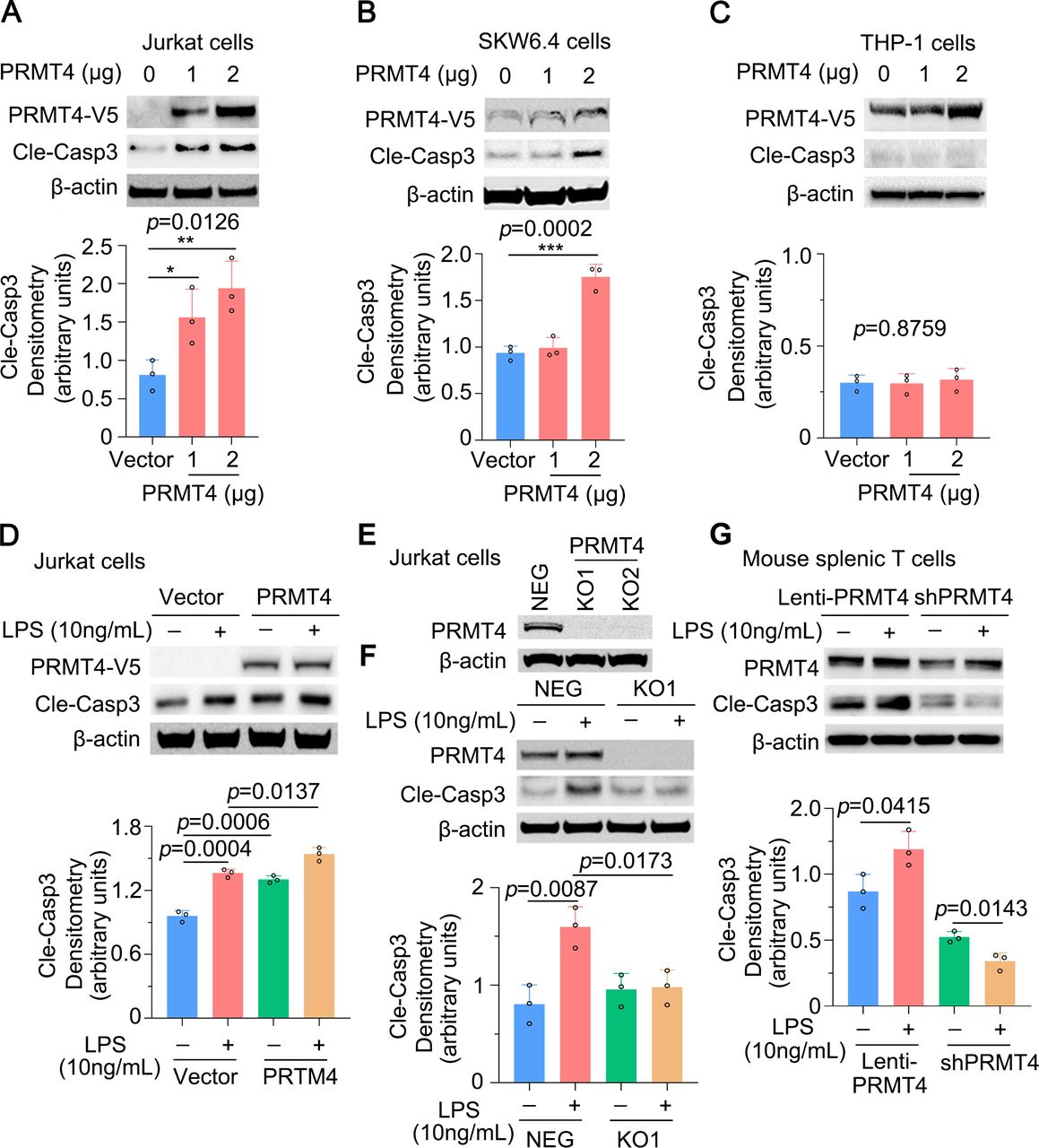
Caspase 3 activation is PRMT4 dependent in lymphocytes. (A,B) Overexpression of PRMT4 increased cleaved caspase 3 baseline levels in Jurkat cells (A) and SKW6.4 cells (B). Relative expression of cleaved caspase 3 was plotted in the lower panel. (C) PRMT4 overexpression does not activate caspase 3 in THP-1 cells. (D) Ectopic expression of PRMT4 enhances LPS-induced caspase 3 activation in Jurkat cells. (E) KO of PRMT4 in Jurkat cells with the CRISPR/Cas9 technique. (F) KO of PRMT4 limits LPS-induced caspase 3 activation. (G) Lentiviral expression of PRMT4 enhances LPS-mediated caspase 3 activation and depletion of PRMT4 by lenti-shPRMT4 reduces cleaved caspase 3 in mouse splenic lymphocytes. Independent experiments, n=3. *P=0.05–0.01, **P=0.01–0.001, ***P=0.001–0.0001. KO, knockout; LPS, lipopolysaccharide; neg, negative; PRMT4, protein arginine N-methyltransferase 4.
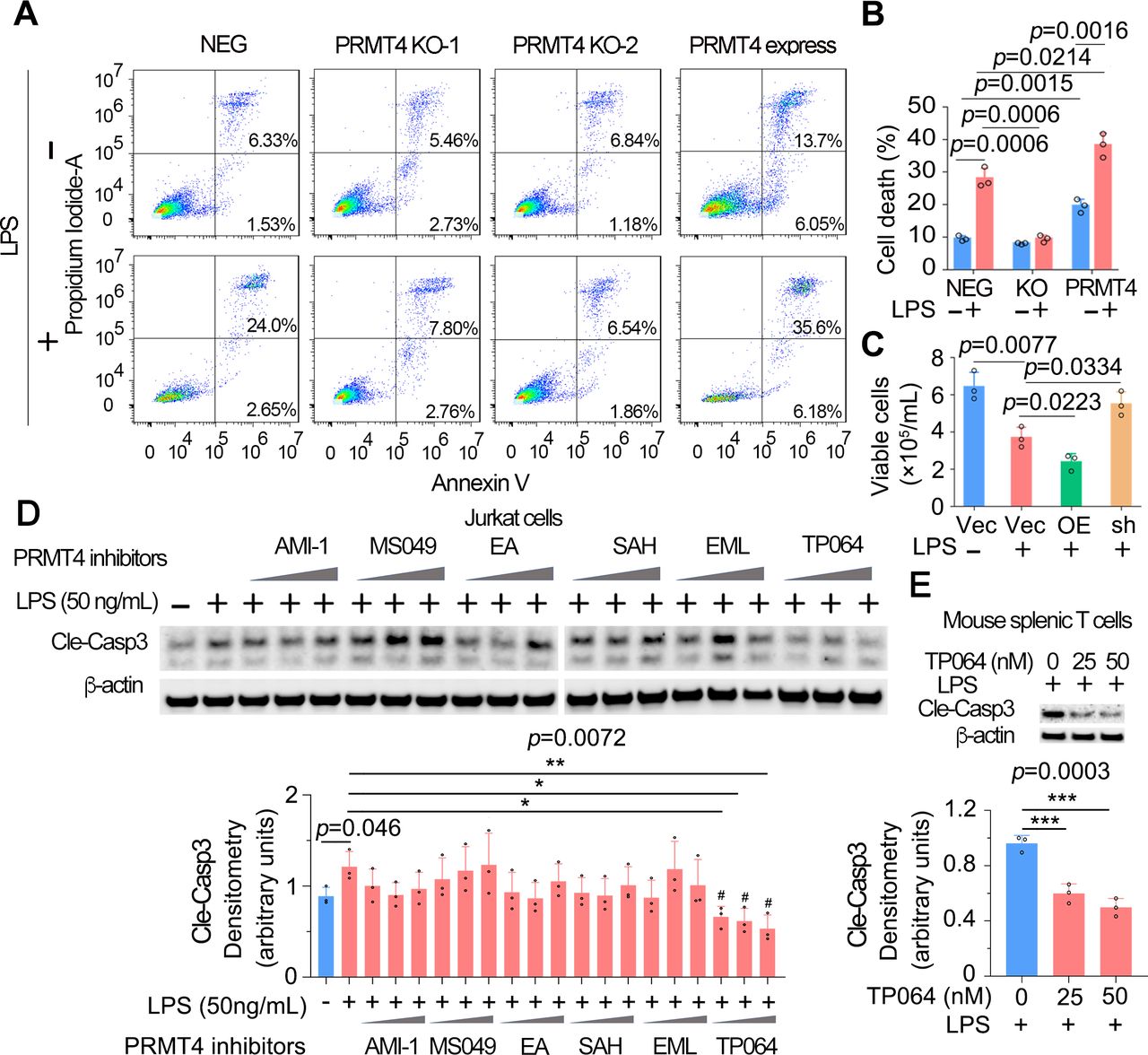
PRMT4 mediates lymphocyte death and PRMT4 depletion or chemical inhibition reduces lymphocyte cell death
To explore the pathophysiological function of LPS-mediated PRMT4 elevation and subsequent caspase 3 activation, we measured lymphocyte apoptosis. We conducted annexin V flow cytometry to confirm the role of PRMT4 in LPS-mediated T-cell death. Flow-cytometry results showed that the baseline levels of cell death were similar in PRMT4 KO Jurkat cells (8.19% and 8.03%) as compared with those of control cells (7.86%). LPS treatment substantially promoted cell death in non-targeted cells (26.65%) but not in PRMT4 KO cells (10.47% and 8.31%). Interestingly, PRMT4 overexpression promoted Jurkat cell death (19.75%) and augmented LPS-induced cell death (41.78%) (figure 5A,B). In mouse splenic T cells, LPS did not affect cell survival in PRMT4 depletion but remarkably reduced cell survival in PRMT4 overexpression (figure 5C). Next, we assessed if a PRMT4 small molecule inhibitors might suppress T-cell death. We first screened commercially available PRMT4 inhibitors in LPS-treated Jurkat cells to test if the inhibitor might inhibit caspase 3 activation. A selective and cell active PRMT4 inhibitor TP064 efficiently and most consistently inhibited caspase 3 activation at a concentration of ~40 nM (figure 5D). Further, experimental results suggest that TP064 can block LPS-mediated caspase 3 activation at both 25 and 50 nM in mouse splenic T cells (figure 5E). These studies link PRMT4 to endotoxin-induced lymphocyte death and showcase the ability of a PRMT4-specific small molecule inhibitor to effectively antagonise endotoxin-induced lymphocyte death signalling.
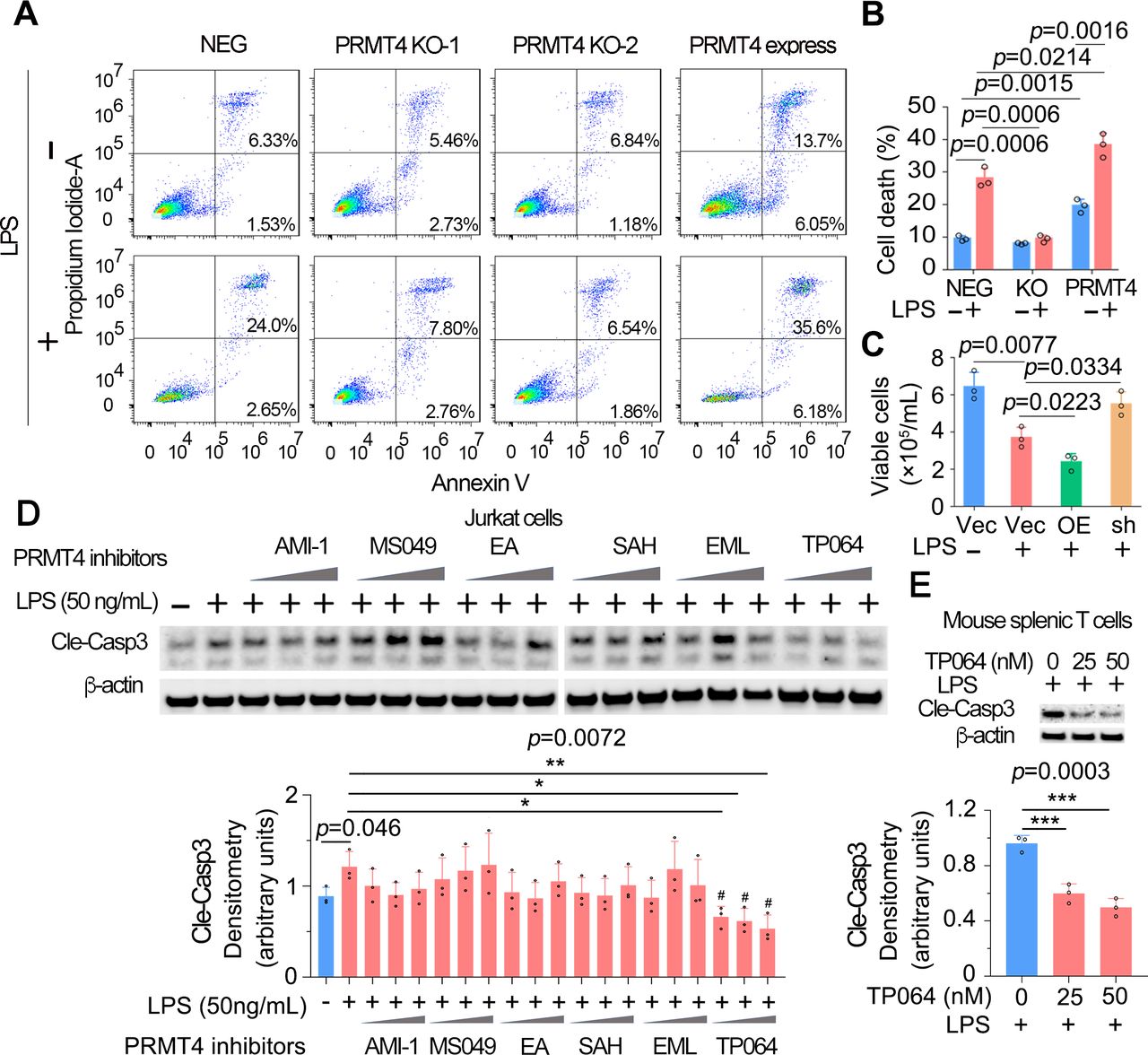
High protein level of PRMT4 causes lymphocyte death. (A,B) FACS analysis of apoptosis in PRMT4 KO or overexpressed Jurkat cells with or without LPS treatment. Data of (A) were quantitated in (B). (C) Lenti-PRMT4 or shRNA particles were delivered intratracheally into the mouse. Mouse splenic T cells were isolated and treated with LPS for 18 hours, and viable cells were counted. (D) Jurkat cells were treated with LPS and a range of PRMT4 inhibitors as indicated for 3 hours. Cell lysates were analysed for cleaved caspase 3. Relative expression of cleaved caspase 3 in each group is plotted in the lower panel. Independent experiments, n=3. (E) Isolated mouse splenic T cells were treated with LPS and TP064; cleaved caspase 3 was immunoblotting analysed and plotted in the lower panel. Independent experiments, n=3. *P=0.05–0.01, *P=0.01–0.001, ***P=0.001–0.0001. FACS, fluorescence-activated cell sorting; KO, knockout; LPS, lipopolysaccharide; neg, negative; OE, PRMT4 overexpression; PRMT4, protein arginine N-methyltransferase 4; sh, PRMT4 shRNA; Vec, vector.
PRMT4 mediates splenic lymphocyte death in experimental LPS injury mice model
To test our hypothesis in animal models, we assessed PRMT4 in an LPS injury mouse model. We overexpressed PRMT4 or knocked down PRMT4 using lentiviral constructs (1×107 cfu/mouse, intratracheal administration) in mice (C57BL/6J) for 14 days. LPS (5 mg/kg, intratracheal in PBS buffer) was then administrated (intratracheal) into mice for 24 hours. As intratracheal administration of LPS induces systemic responses and disseminates to major organs such as the spleen, we observed that LPS induced substantial cell death in the lymphoid white pulp of the spleen, which is rich in both T and B lymphocytes, by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (figure 6A,B). Further, overexpression of PRMT4 markedly enhanced LPS-induced lymphocyte death, whereas silencing of PRMT4 by shRNA significantly diminished cell death. TP064 partially protected splenic lymphocytes from death. We further analysed splenocyte death by flow cytometry and identified that overexpression of PRMT4 enhanced cell death of CD4+ T cells (figure 6C,D). Further, depletion of PRMT4 using shRNA or inhibition of PRMT4 with small molecule decreased CD4+ cell death. We observed similar results in CD8a+ cells and CD45R+ cells (online supplemental figure 1). In addition, results from survival studies indicated that PRMT4 overexpressed mice showed a significantly lower survival rate. Depletion of PRMT4 by shRNA or application of TP064 remarkably improved mouse survival (figure 6E). We obtained similar results in an independent replication study (online supplemental figure 2). Meta-analysis was conducted to analyse the reproductivity using these two sets of independent experiments (figure 6F,G, and online supplemental table 1). Meta-analysis using random-effects REML model showed that overexpression of PRMT4 did not remarkably change the survival in experiments as compared with that of LPS only (figure 6F). TP064 effectively promoted mouse survival in experiments (p=0.03) (figure 6G). However, due to the insufficient sample size in shPRMT4 groups, we were unable to analyse the significance. We obtained similar results by combining the two experiments into one (online supplemental table 1). These data suggest that LPS-enhanced PRMT4 protein expression promotes lymphocyte death and targeting PRMT4 improves survival in an LPS mouse lung injury model.
Supplemental material

Inhibition of PRMT4 suppresses splenic lymphocyte death in an LPS challenged mouse model. (A) PRMT4 was knocked down or overexpressed by IT administrated lentiviral constructs for 14 D. LPS or PRMT4 inhibitor were given intratracheally) as indicated for 24 hours (n=8). Spleen tissues were stained with TUNEL. (B) TUNEL-positive cells in spleen tissues were quantitated. (C,D) CD4+ lymphocytes were isolated from splenic tissues in aforementioned PRMT4 knockdown or overexpression experiments (A) and analysed with flow cytometry. CD4 was used as a T-cell marker. Percentage of apoptosis was quantitated in (D) (n=3). (E) Survival studies were conducted in the LPS lung injury model; mice were observed for 48 hours (n=10). (F,G) Two-stage meta-analysis was conducted using two independent sets of murine data using LPS-only group as reference: PRMT4+LPS (F) and TP064+LPS (G). The data of shPRMT4 group are not shown because the HR was not computable. Two independent experiments were conducted (n=26, (10, 16)). Scale bar=100 µm. LPS, lipopolysaccharide; PRMT4, protein arginine N-methyltransferase 4.
PRMT4 mediates splenic lymphocyte death in a polymicrobial sepsis model
We next tested our hypothesis in a well characterised polymicrobial sepsis mouse model, CLP. Mice were subjected to lenti-PRMT4 overexpression and lenti-shRNA as previously mentioned; CLP procedure was followed to induce sepsis in animals for 48 hours. One group of mice receiving lenti-vector only were given TP064 (0.2 µg/mouse) intravenously following CLP. Consistent with aforementioned observations, results from TUNEL staining of the splenic tissues showed that CLP was sufficient to cause splenic T-cell death in the white pulp (figure 7A,B). Flow cytometric data confirmed these observations (figure 7C,D). Moreover, PRMT4 overexpression mediated splenic CD4+ T-cell death in this model, whereas silencing of PRMT4 by shRNA or administration of TP064 attenuated splenic T-cell death. Consistent results were obtained in CD8a+ T cells and CD45R+ B cells (online supplemental figure 3). Survival studies showed that lenti-PRMT4 overexpression decreased mouse survival, and depletion of PRMT4 by shRNA or application of the PRMT4 chemical inhibitor protected mice from death (figure 7E). We obtained similar results in an independent replication study (online supplemental figure 4). Similar results were obtained from meta-analysis using these two sets of independent experiments (figure 7F–H, online supplemental table 2). Meta-analysis using random-effects REML model showed that overexpression of PRMT4 did not remarkably change the survival in experiments (figure 7F). Depletion of PRMT4 by shRNA significantly promoted survival (p=0.01) (figure 7G). PRMT4 inhibitor TP064 effectively promoted mouse survival as well as compared with that of CLP only (p=0.03) (figure 7H). We obtained similar results by combining the two experiments into one in meta-analysis (online supplemental table 2). In summary, these results suggest that PRMT4 is a critical mediator of lymphocyte death in septic models.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inhibition of PRMT4 suppresses splenic lymphocyte death in a polymicrobial sepsis model. (A) CP was performed in PRMT4 knocked down or overexpressed mice (n=8). TP064 (0.2 µg/mouse) was administrated intravenously in one group for 48 hours. Spleen tissues were stained with TUNEL. (B) TUNEL-positive cells in spleen tissues. (C,D) Isolated splenic CD4+ T cells were analysed by flow cytometry (C). CD4 was used as a T-cell marker. The data from (C) are plotted in (D). (E) Survival studies were conducted in the CLP model, and mice were observed for 5 days (n=16). (F–H) Meta-analysis was conducted among two independent sets of murine data using CLP only as reference group: PRMT4+CLP (F), shPRMT4+CLP (G) and TP064+CLP (H). Two independent experiments were conducted (n=26, (10, 16)). Scale bar=100 µm. CLP, cecal ligation and puncture; PRMT4, protein arginine N-methyltransferase 4.
Discussion
There is mounting evidence that a mechanistic centrepiece of sepsis is immunosuppression from loss of effector lymphocytes, underscoring an unmet need to identify new molecular targets for therapeutic intervention.31 The fundamental new findings in this study are that (1) PRMT4 protein is elevated in the plasma of human subjects with sepsis, preclinical models of sepsis, and lymphocytes exposed to bacterial endotoxin; (2) LPS augments PRMT4 that mediates lymphocyte apoptosis; and (3) chemical inhibition or genetic depletion of PRMT4 attenuates lymphocyte death and protects mice after endotoxin-induced lung injury and polymicrobial sepsis. PRMT4 is aberrantly expressed in breast, prostate and colorectal cancers, and is associated with a poor prognosis by promoting tumour progress and cancer metastasis.32–35 Consistent with these observations, we identified that PRMT4 expression is increased in sepsis. PRMT4 protein was upregulated in cellular and mouse septic models, human infected lung tissue samples and peripheral blood leucocytes from septic patients. Increased PRMT4 protein caused lymphocyte apoptosis, and this finding may be one mechanism underlying the pathogenesis of septic immunosuppression, as apoptosis of lymphocytes is a key feature of sepsis-induced immunosuppression,36 and lymphopenia that develops during sepsis predicts early and late mortality.37
Our studies uncover a unique role for PRMT4 in cell death linked to sepsis. Increased cell death has long been noticed as a key pathway in sepsis-induced immunosuppression, but the factors leading to this increased cell death have not been fully defined.38 39 Our findings here add a novel mechanism of immunosuppression at the epigenetic level: a pathogen-derived product induces cellular concentrations of a chromatin modulator, PRMT4, that in turn mediates lymphocyte death via caspase 3-related apoptosis, thus serving as a potential modulator in sepsis. Interestingly, PRMT4 appeared to cause cell death specifically at the white pulp of the spleen. The spleen white pulp is a lymphatic tissue where both B and T lymphocytes reside. Distinct from the matured lymphocytes in the red pulp, the lymphocytes in the white pulp are mostly naïve cells or cells in lineage differentiation and maturing. Considering CD4+ and CD8+ cells are less in PRMT4 KO mice, our study suggests aberrant PRMT4 protein may play an important role in lineage differentiation for both B and T cells. Further studies are required to elucidate the molecular mechanisms whereby PRMT4 regulates apoptosis, epigenetically or via its methylation function driving post-translational modifications of key regulatory proteins that dictate cellular life span. Recent mass spectrometric studies uncovered more than 130 protein substrates modified by PRMT4 in breast cancer cell lines.40 These targets might be suitable candidates for interrogation of post-translational modifications by PRMT4 associated with T-cell immunosuppression.
We observed higher plasma PRMT4 levels in septic patients compared with non-septic patients. Our results suggest that PRMT4 is highly expressed in leucocytes isolated from septic patients, and PRMT4 is detectable in mouse sera and human plasma. Considering the fact that high levels of PRMT4 are cytotoxic, we speculate that PRMT4 in plasma of septic patients may originate from dying or dead cells. Peripheral blood leucocytes are classified into granulocytes and mononuclear agranulocytes, in which agranulocytes are composed of 35% in terms of total leucocytes. Agranulocytes are mononuclear and can be divided into lymphocytes, monocytes and natural killer cells. We show that LPS increases PRMT4 expression in lymphocyte and monocytes. However, PRMT4 mediates caspase 3 cleavage in lymphocytes but not in monocytes. It remains to be determined if LPS promotes PRMT4 expression and activates caspase 3 in granulocytes. Interestingly, inhibition of PRMT4 by shRNA or with the small molecule inhibitor improved mice survival in both the LPS lung injury and CLP mouse models, suggesting that lymphocyte death may be a critical factor underlying the pathogenesis of endotoxin-mediated injury. We repeated the animal experiments as independent data and analysed the data sets using meta-analysis approaches. Meta-analysis combined two studies together that may increase the power of the experiments, reach more accurate estimate of effect magnitude and strengthen the conclusions from single study. Independent murine experiments may introduce heterogeneity, though the experimental individuals are with high consistence but less heterogeneity. However, meta-analysis is resource wasting and time consuming, and requires experienced statistician to perform the analysis. In addition, not all studies provide adequate data for inclusion and analysis. Overall, pharmaceutical targeting of PRMT4 may be a potential alternative therapeutic approach in septic patients with significantly low T-cell counts and immunosuppression.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by The University of Pittsburgh Institutional Review Board, which also approved the parent Acute Lung Injury Registry and Biospecimen Repository (PRO10110387) and the exempt protocol to use deidentified samples and data for this study (MOD201800140). Participants gave informed consent to participate in the study before taking part. All animal protocols and procedures were reviewed and approved by the University of Pittsburgh Institutional Animal Care and Use Committee (IACUC protocol # IS00010084).
Acknowledgments
We thank Drs Prabir Ray and Anuradha Ray for the in-depth discussion on science.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @KitsiosMd
YL, XiuL, TL, RKM and CZ contributed equally.
Correction notice This article has been corrected since it was first published. The open access licence has been updated to CC BY.
Contributors CZ conceived the science and designed the experiments. CZ serves as the guarantor of the study. YL, XiaoL and TL conducted the immunoblotting analysis. YL and XiuL performed animal studies. KC conducted single-cell RNA sequencing experiments. BJM, XiaL, YZ, MN and GK provided human samples and conducted human-related studies. MN performed the statistical analysis. JSL and RKM helped to develop the science and interpreted the results. The manuscript was written by YL and CZ and edited by TN, YZ, BJM, GK, MN, JSL and RKM. All authors read and approved the final manuscript.
Funding This work was supported by the National Institutes of Health (R01 grants HL125435 and HL142997(CZ); HL096376, HL097376, HL098174, HL081784 and P01 HL114453 (RKM); HL136143, HL142084, HL143285 and P01 HL114453 (JSL); and K23 HL139987 (GK)) and the US Department of Veterans Affairs (grants 5I01CX000105-06 and CX001048 (TN)).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.