Article Text

Abstract
Cellular senescence contributes to the pathophysiology of chronic obstructive pulmonary disease (COPD) and cardiovascular disease. Using endothelial colony-forming-cells (ECFC), we have demonstrated accelerated senescence in smokers and patients with COPD compared with non-smokers. Subgroup analysis suggests that ECFC from patients with COPD on inhaled corticosteroids (ICS) (n=14; eight on ICS) exhibited significantly reduced senescence (Senescence-associated-beta galactosidase activity, p21CIP1), markers of DNA damage response (DDR) and IFN-γ-inducible-protein-10 compared with patients with COPD not on ICS. In vitro studies using human-umbilical-vein-endothelial-cells showed a protective effect of ICS on the DDR, senescence and apoptosis caused by oxidative stress, suggesting a protective molecular mechanism of action of corticosteroids on endothelium.
- COPD pharmacology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Cellular senescence is a fundamental mechanism that contributes to the pathophysiology of age-related disorders, including cardiovascular disease (CVD) and chronic obstructive pulmonary disease (COPD).1 The DNA damage response (DDR) activated by oxidative-stress results in cell cycle arrest, senescence or apoptosis. Senescent endothelial cells are dysfunctional, exhibit a proinflammatory ‘senescence-associated secretory phenotype’ (SASP), promoting vascular inflammation, atherogenesis and thrombosis. Using circulating endothelial progenitors named endothelial colony-forming-cells (ECFC) or blood-outgrowth-endothelial-cells,2 we demonstrated accelerated endothelial senescence in smokers and patients with COPD due to epigenetic dysfunction, supporting the concept of accelerated ageing of the endothelium as a contributor to CVD.3 4
Inhaled corticosteroids (ICS) are widely used in COPD in patients with severe disease and frequent exacerbations. ICS may have a protective effect on cardiovascular comorbidities in COPD, even though this has been controversial and the mechanism is unknown.5 Here, we demonstrate that ICS reduce senescence and SASP in ECFC from patients with COPD, suggesting a novel protective mechanism of action of corticosteroids on endothelium.
Methods
ECFCs were isolated from participants of previous study3 and two newly recruited patients with COPD, as described.3 Informed consent was obtained from all individuals (table 1). Please see online supplemental material for detailed methodology.
Supplemental material
Clinical details of participants
Results
As previously shown, ECFC from healthy smokers and patients with COPD displayed increased senescence and markers of DDR compared with healthy non-smokers as measured by Senescence-associated β-galactosidase (SA-β-gal) activity, p21CIP1, p16INK4, 53BP1 and γ-H2AX ((figure 1A) and in published cohort).3 An unexpected finding from subgroup analysis was that ECFC from patients with COPD on ICS exhibited reduced senescence compared with patients with COPD not on ICS (n=6 COPD-no ICS vs n=8 COPD-ICS) (figure 1A). Reduced senescence in the COPD group on ICS was further confirmed by additional markers of senescence such as p21CIP1 (mRNA (n=6 COPD-no ICS vs n=5 COPD-ICS), immunoblot (n=3 per group), immunofluorescence (n=3 COPD-no ICS vs n=4 COPD-ICS)) and p16INK4 (n=2 per group) (figure 1B–D). We next studied DDR signalling. Mediators of DNA repair are γ-H2AX and 53BP1 that regulate downstream effectors, promoting senescence and apoptosis. We observed reduced markers of DDR in the COPD group on ICS (n=3 per group) (figure 1E), suggesting a protective effect of corticosteroids against DDR and endothelial senescence.
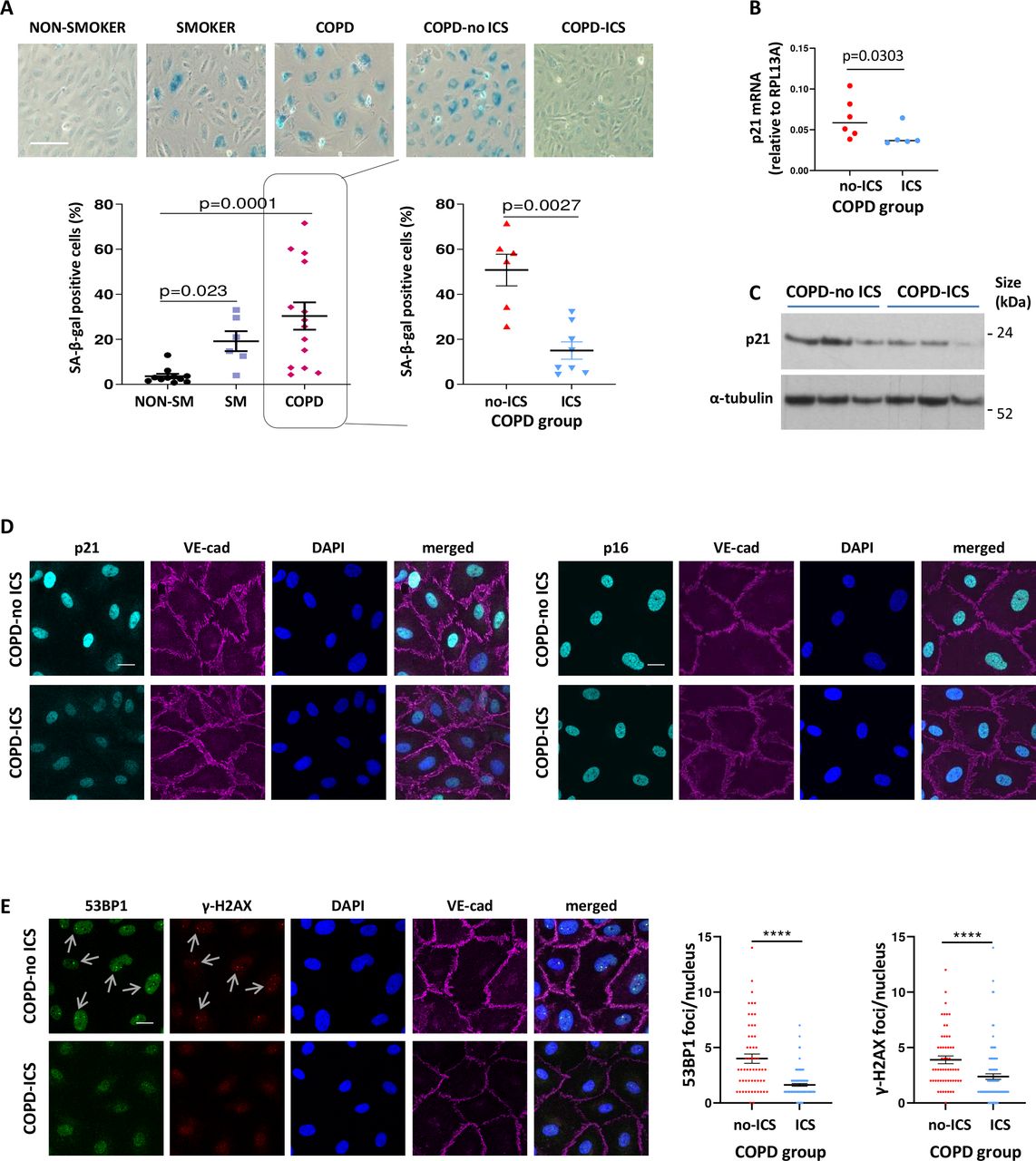
Reduced senescence and DNA damage response in patients with COPD on ICS). (A) Senescence-associated-β-galactosidase activity was assessed as a marker of cellular senescence in ECFC samples from healthy non-smokers (n=11), healthy smokers (n=6) and patients with COPD (n=14, 8 on ICS). ECFC from smokers and patients with COPD exhibited increased senescence compared with non-smokers; Kruskal-Wallis test followed by Dunn’s multiple comparison test (graph A - left panel). ECFC from patients with COPD on ICS exhibited reduced senescence compared with ECFC from patients with COPD not receiving ICS; Mann-Whitney U test (graph A - right panel); (scale bars 100 µm). (B) mRNA levels for p21 were measured by real-time PCR in ECFC from patients with COPD (n=11, 5 on ICS). Ribosomal protein L13a was used for normalisation. (C) p21 protein levels were quantified by Western blotting. α-tubulin was measured for normalisation (n=3 in each group). (D) Representative images of immunofluorescence staining of ECFC from COPD-ICS versus COPD-no ICS patients for p21 (cyan, left panel) and p16 (cyan, right panels). DAPI (blue) was used as nuclear staining and VE-cadherin (magenta) as an endothelial marker. (E) DNA damage was assessed by immunofluorescence staining for 53BP1 (green) and γ-H2AX (red) (n=3 in each group). DAPI (blue) was used as a nuclear marker and VE-cadherin (magenta) as an endothelial marker. The number of distinct nuclear immunofluorescent foci (see arrows) per nucleus was counted in at least 5 z-stack images and 20 cells, using a 63 × objective lens (scale bars = 20 µm). Mann-Whitney U test; ****P<0.0001; COPD, chronic obstructive pulmonary disease; ECFC, endothelial colony-forming cells; ICS, inhaled corticosteroids.
To investigate the possible protective effect of corticosteroids, we performed in vitro experiments on human-umbilical-vein-endothelial cells (HUVEC) cultured under oxidant conditions to induce stress-induced premature senescence (SIPS) in the presence or absence of increasing doses of the ICS budesonide, using three different pooled HUVEC samples. Treatment with budesonide using relevant therapeutic doses of the drug (10−8−10−6mol/L), inhibited SIPS (figure 2A), apoptosis and markers of DDR caused by oxidative-stress (figure 2B,C). We also studied 53BP1 recruitment to sites of DNA damage, appearing by immunofluorescence as distinct nuclear foci caused by oxidative stress, at different timepoints. Budesonide treatment resulted in a reduced number of cells with a high number of foci, and a reduced number of cells with 53BP1 foci compared with controls (figure 2D), further supporting the protective effect of budesonide against oxidative-stress induced DNA damage.

{kind=link}
{kind=link}
Corticosteroids may exert a protective effect against premature endothelial senescence caused by oxidative stress—reduced senescence associated secretory phenotype involving Interferon-gamma (IFN-γ) inducible-protein-10 (IP-10) in patients with chronic obstructive pulmonary disease (COPD) on inhaled corticosteroids. (A) Human-umbilical-vein-endothelial cells (HUVECs) were cultured in the presence or absence of increasing doses of budesonide (10−10−10−6mol/L) or control vehicle (DMSO). Following 1-hour pretreatment, HUVECs were exposed to 50 µM of H2O2 for 1.5 hours to induce stress-induced premature senescence. SA-β-gal activity was measured after 72 hours from H2O2 treatment; n=3 (scale bars 100 µm). (B) Apoptosis was quantified by measuring caspase-3/7 Glo activity after 24 hours from H2O2 treatment as described in (A); n=3 (samples in triplicate). (C) γ-H2AX protein after 24 hours from H2O2 treatment as described in (A); n=3. (A–C) Friedman test followed by Dunn’s multiple comparison test. (D) HUVECs were stained for 53BP1 and for DRAQ5 (nuclear marker) at 60 min, 240 min, 24 hours or 48 hours after exposure to H2O2 treatment. The number of 53BP1 positive cells and the number of foci per cell were quantified (scale bars 20 µm). (E) IL-8 and IFN-γ-inducible protein 10 (IP-10 or CXCL10) were measured in supernatant samples from ECFC cultures under baseline conditions from non-smokers (n=5) and patients with COPD (n=8; n=5 COPD-ICS) by a Luminex assay. Pearson correlation coefficient of IL-8 and IP-10 with SA-b-gal activity. Reduced expression of IP-10 was observed in samples from patients with COPD on ICS compared with patients with COPD not receiving ICS; Kruskal-Wallis test followed by Dunn’s multiple comparison test. (F) Immunofluorescence staining for IP-10 (green) and p21 (cyan). DAPI (blue) was used as a nuclear marker and VE-cadherin (magenta) as an endothelial marker. At least 5 z-stack images and 20 cells per ECFC sample were analysed for IP-10 and p21 using a 63 × objective lens in ECFC from COPD-ICS (n=4) and COPD-no ICS (n=3) (scale bars = 20 µm); Mann-Whitney U test; ****p<0.0001. DMSO, Dimethyl sulfoxide; ECFC, endothelial colony forming cells; ICS, inhaled corticosteroids; IL-8, interleukin-8.
Chemokines released from endothelial cells promote vascular inflammation. We measured 22 proinflammatory cytokines in ECFC supernatant collected under baseline conditions from non-smokers (n=5) and patients with COPD (n=8) receiving ICS or not. Cytokines included key SASP components such as interleukin (IL)-1α, IL-6, IL-8 and Interferon-gamma (IFN-γ)-inducible-protein-10 (IP-10). We found a positive correlation between IL-8 and SA-β-gal (Pearson’s r=0.6774, 95% CI 0.167 to 0.899, p=0.017), and IP-10 and SA-β-gal (Pearson’s r=0.6998, 95% CI 0.210 to 0.909, p=0.011) (figure 2E), suggesting that IL-8 and IP-10 constitute part of the SASP in ECFC. Intriguingly, IP-10 levels in culture supernatant were reduced in patients with COPD on ICS compared with those who were not, a finding that was also confirmed when studying IP-10 expression directly in ECFC by immunofluorescence (n=3 COPD-no ICS vs n=4 COPD-ICS). Both intracellular and nuclear expression of IP-10 was reduced in the samples from COPD on ICS (figure 2F). These results suggest a beneficial effect of ICS on the ECFC secretory phenotype involving IP-10.
Discussion
We demonstrated that endothelial cells from patients with COPD showed increased senescence and SASP, which may be modified by ICS. The effect of corticosteroids on vascular ageing has not been extensively investigated. Corticosteroids appear to have beneficial or detrimental effects on the vasculature depending on the context. The glucocorticoid-receptor is ubiquitously expressed on endothelial cells and is a negative regulator of vascular inflammation.6 In COPD, evidence suggests a protective effect of ICS on cardiovascular comorbidities, which is further supported by the recent IMPACT (Informing the Pathway of COPD Treatment) and ETHOS (Efficacy and Safety of Triple Therapy in Obstructive Lung Disease) trials, prospectively demonstrating reduced mortality (including from CVD) in patients with COPD treated with ICS, including budesonide.7 In this study, we demonstrate that patients with COPD on ICS exhibit significantly reduced endothelial senescence and IP-10 release compared with patients with COPD not on ICS. These findings were reflected in in vitro experiments using budesonide, which reduced DDR, premature senescence and apoptosis caused by oxidative-stress, suggesting a novel and protective molecular mechanism of action of corticosteroids on endothelium.
IP-10 functions as a leucocyte chemoattractant and promotes endothelial senescence and atherogenesis. Interestingly in the current COVID-19 pandemic, IP-10 is a biomarker of severity and possibly contributes to the pathophysiology of severe disease.8 Corticosteroids are beneficial in COVID-19 patients with respiratory failure,9 and ICS in symptomatic patients.10 We can, therefore, speculate a protective effect of corticosteroids against endothelial senescence and inflammation, promoting vascular homeostasis and integrity, important for cardiovascular comorbidities and possibly for severe complications in COVID-19.
Our study is mainly retrospective involving a small number of patients, and mechanistic findings have been confirmed only with budesonide. The protective effect of corticosteroids against endothelial senescence was observed in patients with COPD that were on different ICS, suggesting that this effect is applicable to multiple ICS. Future prospective clinical and mechanism studies are required to investigate the relationship between glucocorticoid and IFN‐γ‐mediated pathways in the context of vascular ageing and confirm the suggested beneficial effect of ICS on the endothelium. If this is the case, ICS may protect patients with COPD and other groups characterised by endothelial senescence (eg, smokers) from cardiovascular comorbidities, and from endothelial driven complications in viral diseases.
Ethics statements
Patient consent for publication
Ethics approval
This study was approved by South East Scotland Ethics Committee 02 (ID: 284894 REC reference: 20/SS/0085), Royal Marsden, Hammersmith and Queen Charlotte’s Ethics Committees.
Acknowledgments
We thank Mrs Sally Meah for collecting patients' samples, and Dr Richard Starke and Dr Graeme Birdsey for scientific and technical advice (Imperial College London). We also thank the Facility for Imaging by Light Microscopy (FILM) at Imperial College London. We apologise in advance for omitting references and reviews due to space limitations and we provide a list in online supplemental material.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
AMR and PJB are joint senior authors.
CR and CP contributed equally.
Contributors Conception and design: KP, AMR and PJB; Data analysis and interpretation: KP, CP, SR, GCD, JAW, VG, AMR and PJB; Experimental performance: KP, CR, CP, MM and SR; Writing of the manuscript: KP, AMR and PJB.
Funding This work was funded by a Wellcome Trust Programme Grant (093080/Z/10/Z) and AstraZeneca AB Project Grant (WHRD_P37317). KP is financially supported by National Heart & Lung Institute-Imperial College London and a British Heart Foundation Project Grant (PG/19/75/34686). KP was supported by National Institute for Health Research (NIHR) Biomedical Research Centre based at Imperial College Healthcare NHS Trust and Imperial College London.
Competing interests Part of this work was funded by an academic AstraZeneca AB Project Grant.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves