Article Text

Abstract
Background Air pollution exposure is associated with disease severity, progression and mortality in patients with idiopathic pulmonary fibrosis (IPF). Combined impacts of environmental and socioeconomic factors on outcomes in patients with IPF are unknown. The objectives of this study were to characterise the relationships between relative environmental and social disadvantage with clinical outcomes in patients with IPF.
Methods Patients with IPF were identified from a longitudinal database at University of California, San Francisco. Residential addresses were geocoded and linked to the CalEnviroScreen 3.0 (CES), a tool that quantifies environmental burden in California communities, combining population, environmental and pollution vulnerability into individual and composite scores (higher scores indicating greater disadvantage). Unadjusted and adjusted linear and logistic regression and Fine and Gray proportional hazards models were used.
Results 603 patients were included. Higher CES was associated with lower baseline forced vital capacity ( β =−0.073, 95% CI −0.13 to −0.02; p=0.006) and diffusion capacity of the lung for carbon monoxide ( β =−0.11, 95% CI −0.16 to −0.06; p<0.001). Patients in the highest population vulnerability quartile were less likely to be on antifibrotic therapy (OR=0.33; 95% CI 0.18 to 0.60; p=0.001) at time of enrolment, compared with those in the lowest quartile. An association between CES and mortality was suggested, but sensitivity analyses demonstrated inconsistent results. Relative disadvantage of the study cohort appeared lower compared with the general population.
Conclusions Higher environmental exposures and vulnerability were associated with lower baseline lung function and lower antifibrotic use, suggesting that relative socioenvironmental disadvantage has meaningful impacts on patients with IPF.
- interstitial fibrosis
- clinical epidemiology
- idiopathic pulmonary fibrosis
Data availability statement
Data are available on reasonable request.
Statistics from Altmetric.com
Key messages
What is the key question?
Do environmental exposures and population vulnerability impact clinical outcomes in patients with idiopathic pulmonary fibrosis (IPF)?
What is the bottom line?
Relative disadvantage is associated with lower baseline lung function and lower antifibrotic use in patients with IPF.
Why read on?
Understand how environmental exposures and social vulnerability affect patients with IPF, including lung function, antifibrotic use and survival.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic interstitial lung disease (ILD) of complex aetiology. The prognosis associated with IPF remains poor at 3–5 years from diagnosis,1 and disease-attributable mortality rates have increased worldwide.2 3 Environmental risk factors for IPF include a history of cigarette smoking, occupational and inhalational exposures such as dusty work environments and exposure to secondhand smoke.4 Higher exposures to air pollution are associated with risk of developing disease, and worse outcomes in patients with IPF.5 Ozone (O3) and nitrogen dioxide (NO2) exposures are associated with increased risk of acute exacerbations,6 and long-term exposure to high levels of particulate matter (PM) and NO2 are associated with higher risk of death in patients with IPF.7 8 Increased exposure to PM has also been associated with lower forced vital capacity (FVC),9 and accelerated decline in FVC over time.10 Taken together, these data inform relationships between air pollution exposure and clinical outcomes in patients with IPF.
Socioeconomic status (SES) has been associated with health disparities in patients with IPF.7 A US-based study demonstrated that among hospitalised patients with IPF, those with lower SES were less likely to undergo lung transplant as compared with patients in the highest income quartile.11 Another US-based study showed higher mortality in Black and Hispanic IPF patients referred for lung transplantation, compared with white patients, with disparate outcomes attributed to lower SES and systemic racism.12 A recent study by Sesé et al found that low household income was associated with lower survival in patients with IPF, informing the need to characterise social disparities in this patient population.13 Research in environmental inequity, defined as the distribution of exposures across different socioeconomic groups, has demonstrated that environmental exposures contribute to adverse health outcomes more disproportionately in lower SES communities.14 These disparities are attributed to higher pollution, as well as increased susceptibility to adverse environmental exposures.14 The combined impacts of environmental exposures and susceptibility to their adverse effects are recognised risks for poor health outcomes. Higher pollution exposures may reflect social and economic vulnerability, yet to date, no studies have concurrently evaluated these combined impacts, or the cumulative environmental burden, on clinical outcomes in patients with IPF.
The objectives of this study were to characterise the relationships between individual and composite measures of social vulnerability and environmental health with baseline disease severity in patients with IPF, using an established environmental health tool, CalEnviroScreen 3.0 (CES).5 We further sought to characterise the associations of social vulnerability and environmental health with survival and use of antifibrotic therapies in this patient population.
Methods
Study population
Patients with IPF were retrospectively identified from the ILD programme database at the University of California, San Francisco (UCSF), enrolled between 2001 and 2017 (median year 2012). All IPF diagnoses were established according to contemporaneous guidelines1 15 16 via review in a formal multidisciplinary discussion. All patients seen in ILD clinic were eligible for enrolment in the database, with data censored 1 December 2019. Patients required a residential zip code within the state of California to be included in this study. All patients provided written informed consent for database enrolment.
Baseline data collected at time of diagnosis included age, sex, smoking history, residential addresses, FVC % predicted and diffusion capacity of the lung (DLCO) % predicted. All longitudinal pulmonary function tests were obtained from diagnosis to time of censor. Treatment with an IPF-specific antifibrotic medication (pirfenidone or nintedanib), was recorded at time of initial referral for all patients with ongoing follow-up subsequent to 1 November 2014. US Food and Drugs Administration approval for both anti-fibrotic medications occurred 15 October 2014. Dates of death or lung transplantation were recorded, and patients were censored at time of death, lung transplantation or at time of data extraction (1 December 2019). The outcomes of interest were baseline disease severity, rate of FVC% decline, time to death or lung transplantation, and baseline use of antifibrotic medications.
Exposure variables
Residential addresses were geocoded to provide census block group for each patient. Geocoded addresses were matched to census block codes using the Texas A&M geocoding service (https://geoservices.tamu.edu).
Environmental and social metrics for individual patients were based on the CES, which was last updated in 2016.17 The CES is a validated tool, developed by the California Environmental Protection Agency Office of Environmental Health Hazard Assessment. CES identifies California communities by census tract that are disproportionately burdened by, and vulnerable to, multiple sources of pollution.17 The CES characterises community-level pollution burden, as well as vulnerability to pollution exposures based on population characteristics. The CES is assigned by census tract, which allowed for easy assignment to the patient by census block group. CES considers 19 total indicators to characterise community-level pollution burden (12 indicators) and vulnerability, based on population characteristics (7 indicators). Pollution burden includes environmental exposures (O3, PM less than 2.5 μm in aerodynamic diameter (PM2.5), diesel emissions, drinking water contaminants, pesticide use, toxic releases from facilities and traffic densities) and environmental effects (cleanup sites, groundwater threats, hazardous waste, impaired water bodies and solid waste sites). Population characteristics encompass five socioeconomic indicators (educational attainment, low income household, linguistic isolation, poverty and unemployment), and three sensitive populations indicators (asthma, low birth weight and cardiovascular disease). The pollution burden and population characteristics scores are combined into a composite total score, with higher scores representing greater exposures and susceptibility to negative effects of exposures, that is, relative disadvantage. CES individual and composite scores are presented as percentiles for populations across California, with percentiles used as the independent variables in the current analyses. Given published associations between specific pollutants and IPF, the independent variables were determined a priori to include the composite CES, pollution burden, population characteristics, O3 and PM2.5 percentiles.
Statistical analysis
Means and SD or medians and IQRs were used to describe the study population. Linear regression was used to describe associations between CES scores and baseline characteristics and lung function. Linear mixed models were used to test the relationship between CES scores and change in FVC% over time, in unadjusted analysis and in models adjusted for age, sex and baseline FVC%. A random intercept was included in the linear models. Fine and Gray proportional hazard subdistribution model, an alternative proportional hazard model in the presence of competing events, was used to analyse associations between CES scores and risk of death (with lung transplant as a competing risk), in unadjusted models, and in models adjusted for covariates. Adjusted models included (1) age and sex, (2) age, sex and baseline FVC% and (3) age, sex, baseline FVC% and an interaction term for CES and baseline FVC%.18 Unadjusted cumulative incidence function curves, which estimates the marginal probability for each competing event, were used to compare survival differences between quartiles of CES, and statistically compared using the modified χ2 test statistic proposed by Gray.19 Logistic regression was used to test the probability of being on an antifibrotic therapy as a function of CES scores, comparing across quartiles. Missing values were assumed to be at random and no data were imputed. Analyses were performed using R statistical software V.3.5.2.20 Values of p<0.05 were considered statistically significant.
Results
Study population
A total of 609 patients with IPF were identified from the database. Six patients were excluded due to missing baseline PFT data, with 603 included in the analysis. Baseline characteristics of the cohort are presented in table 1. The mean (±SD) age was 70.5±8.5 years, 75.5% were male, and 70% were ever smokers. Mean baseline FVC% and DLCO% were 69.2±17.7 and 46.3±17.4, respectively. Gender-age-physiology scores21 were calculated for all subjects, with 30% of patients categorised as Stage I, 51.4% stage II, and 17.6% of patients stage III. Median duration of follow-up was 2.2 (IQR 1.9–2.5) years. A median of 3 (IQR 2–7) longitudinal FVC measurements were collected, with a median interval of 2.1 (IQR 1.0–3.8) years between measurements. Of 398 patients with follow-up subsequent to 1 November 2014, 216 patients (54%) were treated with an antifibrotic medication.
Baseline cohort characteristics
Figure 1 presents a map of California with distributions of CES percentiles across the state and within our study population. 86.2% of the patients resided within census blocks containing three or fewer study patients. The mean (±SD) CES percentile was 31.4±28.8 and 53/603 (8.8%) patients lived in disadvantaged communities, defined as the 25% highest scoring census tracts using CES results.17 The distributions of CES, population characteristics, PM2.5, and O3 percentiles for the study population are presented in figure 2A–D. CES percentiles and population characteristics demonstrate similar distributions, which are left-shifted and non-uniform, with over 50% of the study population found in the lower 30th percentiles. Mean exposures for PM2.5 and O3 averaged over 3 years from 2012 to 2014 are presented in table 2. No measures of O3 exceeded current standards of the US EPA National Ambient Air Quality Standards (NAAQS), but 81 patients exceeded the maximum annual PM2.5 NAAQS standard.22 Of these 81 patients, 37 patients were noted to live in disadvantaged communities. PM2.5 and O3 demonstrate bimodal distribution, with 60%–70% of the study population in the lower 30th percentiles, and 15%–20% in the upper 30th percentile.
CalEnviroScreen 3.0 (CES) measures for the study cohort

Map of California representing CES percentiles (A) in general population and (B) in study population. CES, CalEnviroScreen 3.0.
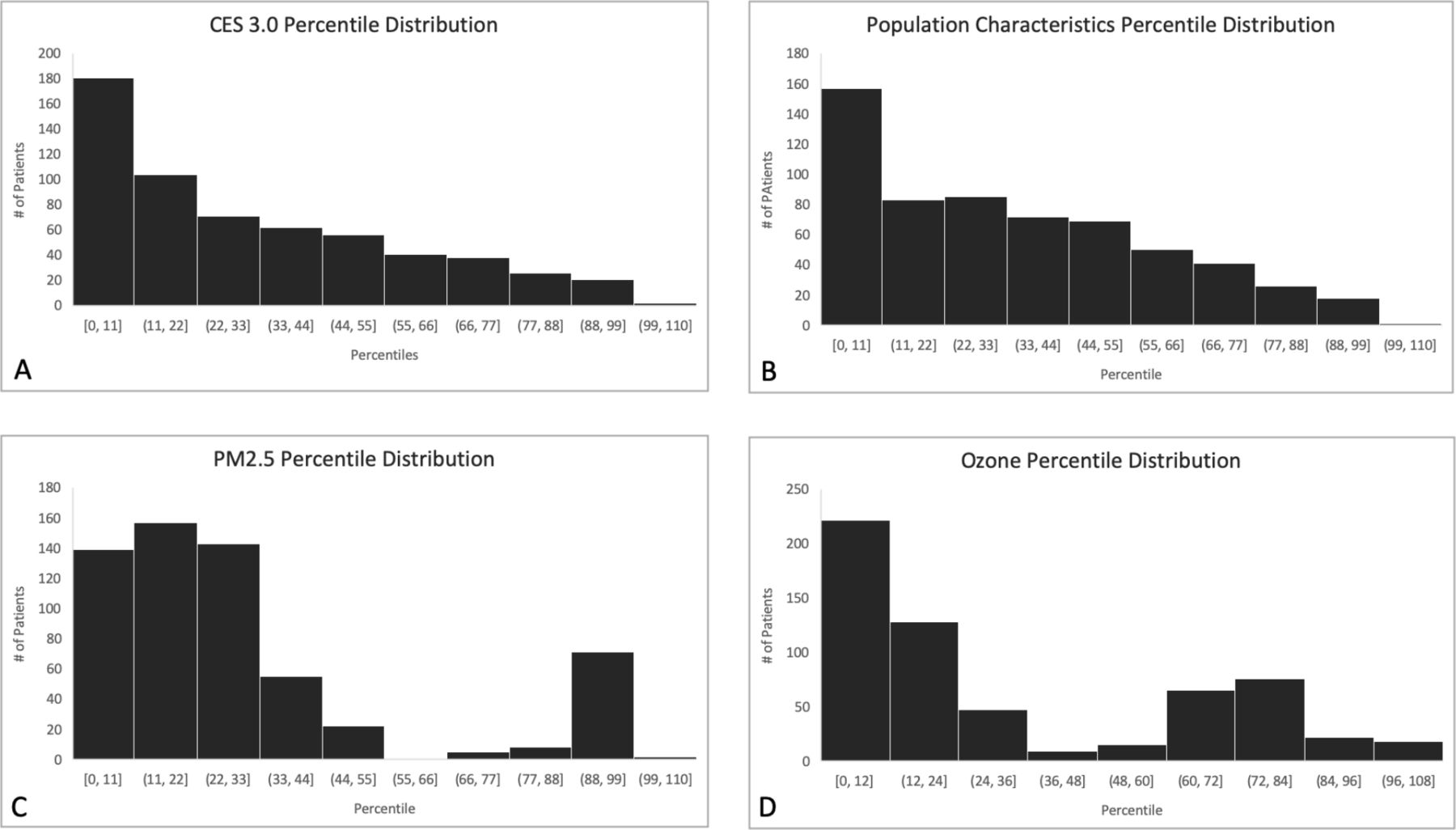
Study population distribution for CES, population characteristics, particulate matter and ozone percentiles. (A) CES percentile distribution. Y axis label=number of patients, X axis label=percentile (B) population characteristics percentile distribution. Y axis label=number of patients, X axis label=percentile (C) (PM)2.5 percentile distribution. Y axis label=number of patients, X axis label=percentile (D) ozone percentile distribution. Y axis label=number of patients, X axis label=percentile. CES, CalEnviroScreen 3.0; PM2.5, particulate matter less than 2.5 μm in aerodynamic diameter.
CES and baseline lung function
Associations between CES percentiles and baseline lung function are presented in table 3. Higher CES percentile was associated with lower baseline FVC% (β=−0.073, 95% CI −0.13 to −0.02; p=0.006) and DLCO% (β=−0.11, 95% CI −0.16 to −0.06; p<0.001). Similarly, higher population characteristics percentile was associated with lower baseline FVC% and DLCO%. Pollution burden was associated with lower baseline DLCO% but not with FVC%. Higher O3 and PM2.5 percentiles were both associated with lower baseline DLCO%, but not with FVC%.
Environmental health measures and baseline lung function
CES and clinical outcomes
The mean annual change in FVC% for the overall cohort was −4.6% /year (SD=13.3). CES quartiles were not associated with annual change in FVC% over time, in adjusted or unadjusted models. We did not identify associations between individual pollutants (PM2.5, O3) or composite pollution burden with annual changes in FVC%. Higher population characteristics quartile was associated with more rapid annual decline in FVC% in unadjusted, but not adjusted models.
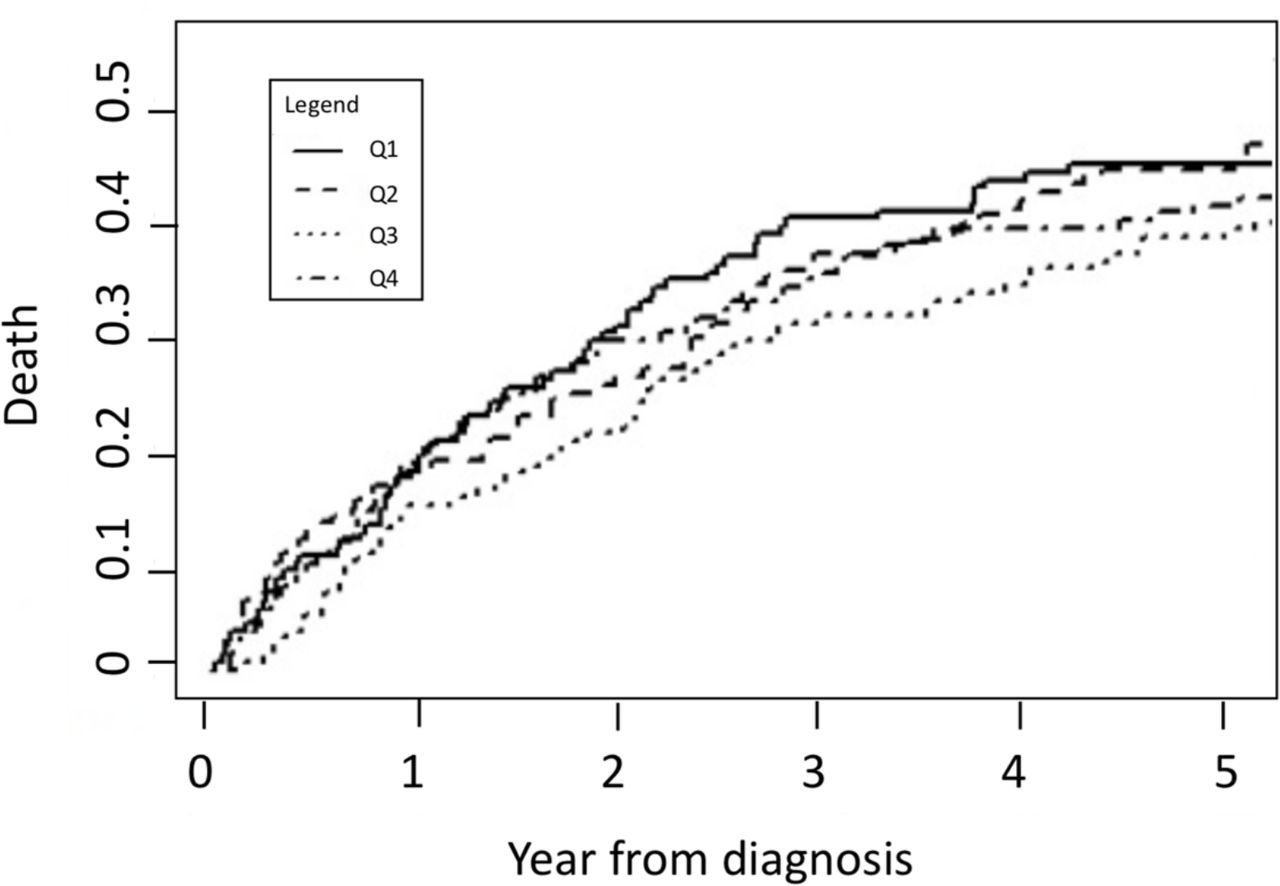
During the follow-up period, 317/603 patients (52.6%) died and 71 patients (11.7%) underwent lung transplantation. We did not identify a consistent relationship between CES and risk of death (figure 3). In unadjusted models, we found no difference in risk of death between quartiles of CES (table 4). Models that were adjusted for age and sex only found no association with death (HR 1.14, 95% CI 0.82 to 1.59; p=0.42). Patients in the highest CES quartile had a lower risk of death compared with the lowest quartile (HR 0.36, 95% CI 0.18 to 0.71; p=0.01), in models adjusted for age, sex and FVC%. In the model that included the CES-FVC% interaction term, the interaction term was significant (HR 1.026, 95% CI 1.007 to 1.046; p=0.009), indicating that despite no overall increased risk of death between highest and lowest CES quartiles, patients in the highest CES quartile were at greater risk of death for a given baseline FVC%, compared with those in the lowest quartile (for marginal means, see online supplemental figure S1). We identified no association between pollution burden with survival in adjusted models (HR 0.99, 95% CI 0.98 to 1.01).
Supplemental material
CES and risk of death with transplant as competing risk
{kind=link}
{kind=link}
{kind=link}
Unadjusted cumulative incidence curves for death in study population.
For patients with follow-up beyond 1 November 2014, CES quartiles were associated with likelihood of receiving antifibrotic medication at time of initial referral, suggesting delayed treatment initiation. Patients in the highest CES quartile were less likely to be on anti-fibrotic therapy (OR 0.39; 95% CI 0.22 to 0.70; p=0.002) at referral, compared with those in the lowest quartile. This finding was seen among highest and lowest population characteristics quartiles (OR 0.33; 95% CI 0.18 to 0.60; p=0.001), but not among pollution burden quartiles (OR 0.65; 95% CI 0.37 to 1.15; p=0.14). We did not identify associations between O3 (OR 0.96; 95% CI 0.63 to 1.46; p=0.86) or PM2.5 quartiles (OR 0.73; 95% CI 0.49 to 1.01; p=0.12) and use of antifibrotic therapy.
Discussion
In this study, we found that higher exposures to environmental pollutants and greater social vulnerability, including both composite and individual CES measures, were associated with lower baseline lung function in patients with IPF. Furthermore, patients living in regions of greater relative disadvantage were less likely to be on antifibrotic therapy at their time of referral to the ILD centre, suggesting delayed treatment initiation despite lower lung function. Previous studies have demonstrated associations between air pollution, lung function and survival in patients IPF,6 7 9 10 but this is the first study to demonstrate combined relationships between SES, environmental health and clinical characteristics in this patient population.
Higher exposures to air pollution have been associated with lung function, and lung function decline across multiple cohorts of IPF patients.9 10 Our findings are consistent with previous reports, and demonstrate a novel association between baseline lung function and CES population characteristics. This metric is a composite of several variables including poverty, unemployment, housing insecurity, education and linguistic isolation, among others.17 The specific reasons behind this finding are unclear, but may be because those in higher population characteristics percentiles received a delayed diagnosis or delays in referral to a tertiary ILD clinic, suggesting that societal inequities contribute to health characteristics in IPF. Greater relative disadvantage, including higher exposures to air pollutants, may have led to these patients having lower baseline lung function, even prior to their diagnoses of IPF. Further studies are required to understand these relationships and to evaluate potential solutions to mitigate differential disease outcomes, and differences impacting baseline lung function in vulnerable groups.
Patients with higher population characteristics and CES scores were less likely to be on antifibrotic therapy at time of referral to the ILD centre, when comparing the highest and lowest quartiles among the study population. Without more granular data, we cannot fully explain this discrepancy. Disease specific therapies may not have been offered to patients in these highest quartiles, patients may have preferred not to take them, or the high cost of these drugs may have been a deterrent. A recent study of claims-based data demonstrated the high out of pocket costs of anti-fibrotic treatment for some patients in the USA, with an average US$430/month for patients on Medicare, and US$150/month for patients covered by commercial insurance.23 This finding appears to be driven by population characteristics rather than pollution exposures, where the relative disadvantage metrics included are surrogates of SES, thus, the discrepant use of antifibrotics in this cohort may reflect inequities in access to guideline recommended pharmacologic treatment, and delays in treatment initiation.24 Longitudinal granular data will help better understand the use of IPF-specific therapies after having been seen at an ILD referral centre, as well as to identify potential barriers to use.
We found inconsistent relationships between CES and risk of death in this study population, which we cannot fully explain. Patients in the highest CES quartile had a lower risk of death compared with lower quartiles in this study population, in models adjusted for age, sex and FVC% despite lower baseline lung function and less use of antifibrotics. This finding was not seen in models adjusted for age and sex only, or models including the interaction term. Patients residing in the more relatively disadvantaged regions were younger at baseline, potentially reducing the impact of the other variables. There was no difference among the quartiles for follow-up duration or date of enrolment (data not shown) therefore this is unlikely due to lead time bias or changing practice patterns over time. Other unmeasured variables may be more strongly impacting survival in this cohort, compared with the burden of environmental exposures measured by the CES, and we could not further characterise comorbidities or other IPF-related complications such as hospitalisation. Interestingly, the significant interaction between CES and FVC% indicates that higher baseline lung function may not be as protective against death in the most vulnerable group, compared with the least vulnerable group. Given the inconsistency in our findings, this complex interplay between relative disadvantage and survival in patients with IPF should be considered in future prognostic models. To ensure generalisability, these should be developed and validated in patients across diverse social and environmental domains.
An unanticipated finding from our data was the non-uniformity of cumulative environmental burden in this cohort, compared with the general population of California. The distribution was left skewed for the CES percentile and its composite measures, as well as PM2.5 and O3. Our study population was obtained from a single centre, and appears to not represent patients with higher CES scores that is, those at greater relative disadvantage. There are several potential explanations, and potential impacts of this finding. The UCSF ILD programme is in San Francisco, one of the most expensive cities in the USA. While it is a referral centre that patients can access from many areas, the clinical cohort may primarily reflect a higher SES demographic from this region. Alternatively, patients with IPF may preferentially move to areas with lower air pollution to avoid triggers for exacerbations or symptoms, or to reside closer to clinics where they receive specialty care, although this seems less likely. Importantly, the non-uniform distribution of relative disadvantage highlights that tertiary referral cohorts may not be reflective of the general population, limiting the generalisability of research findings from such institutions including clinical trials of therapeutics.25 Such non-uniformity may also reflect disparate access to subspecialty ILD care and clinical trial enrolment and participation. However, further work is needed to understand referral patterns and their implications for research generalisability in this field.
Our study has limitations. The patient population was recruited from a single centre and these findings may not be generalisable to other regions or health systems. We did not have data on race or ethnicity, which will be important variables to include in future studies on SES and relative disadvantage. Pulmonary function testing data were obtained from multiple labs, which may have used different norm set values and quality assurance, potentially introducing error; this highlights the importance of standardised interpretation when considering % predicted values. The CES scores, and thus all estimates for environmental exposures and vulnerability were ascribed based on residential zip codes. While this provides data relevant to the domestic environment, it does not account for occupational, avocational or other exposures occurring outside the home. The exposure estimates are made at the neighbourhood level, and we cannot exclude misclassification bias on the individual level. Additional misclassification bias may exist given that patients may have moved over the course of follow-up, but the direction of potential bias (if any) is unclear. The application of community-level exposure variables to individuals may be inaccurate, however without granular data on individual-level SES and environmental exposures, we cannot overcome this potential source of misclassification bias. Finally, we cannot exclude the potential for residual unmeasured confounding.
In summary, higher environmental exposures and social vulnerability, as measured by CES percentiles, were associated with lower baseline lung function and lower baseline antifibrotic use in this cohort of patients. These findings suggest that environmental health and relative disadvantage are associated with clinical characteristics and treatment patterns in patient with IPF. Future studies should aim to characterise the mechanisms underlying these relationships, and to develop strategies to address and mitigate any observed inequities.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by University of Calgary REB 17-078 UCSF REB 10-01592.
Acknowledgments
The authors would like to thank the patients, clinicians and coordinators from the UCSF ILD program for their ongoing commitment to clinical research. We would also like to thank Ms. Jane Berkeley for administrative support for this project.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors NA, EMN, SDN and KAJ conceived of the study. All authors provided critical intellectual input on study design, data analysis and interpretation, and manuscript preparation. KAJ is the acting guarantor and accepts full responsibility for the work.
Funding This study was funded by the CHEST Foundation Research Grant for Pulmonary Fibrosis.
Map disclaimer The depiction of boundaries on the map(s) in this article does not imply the expression of any opinion whatsoever on the part of BMJ (or any member of its group) concerning the legal status of any country, territory, jurisdiction or area or of its authorities. The map(s) are provided without any warranty of any kind, either express or implied.
Competing interests SDN is a consultant and is on the speakers bureau for Roche-Genentech and Boehringer-Ingelheim. He is also a consultant for Bellerophon, United Therapeutics and Galapagos. KAJ reports grants, personal fees and other from Boehringer-Ingelheim, personal fees and other from Hoffman La Roche, personal fees and other from Theravance, personal fees and other from Blade Therapeutics, grants from Chest Foundation, grants from University of Calgary School of Medicine, grants from Pulmonary Fibrosis Society of Calgary, grants and personal fees from Three Lakes Foundation, personal fees from Pliant Therapeutic, outside the submitted work.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.