Article Text

Abstract
Familial spontaneous pneumothorax (FSP) accounts for 10% of primary spontaneous pneumothoraces. Appropriate investigation of FSP enables early diagnosis of serious monogenic diseases and the practice of precision medicine. Here, we show that a pneumothorax genetics multidisciplinary team (MDT) can efficiently diagnose a range of syndromic causes of FSP. A sizeable group (73.6%) of clinically unclassifiable FSPs remains. Using whole genome sequencing we demonstrate that most of these cases are not known monogenic disorders. Therefore, clinico-radiological assessment by an MDT has high sensitivity for currently known clinically important monogenic causes of FSP, which has relevance for the design of efficient pneumothorax services.
- pleural disease
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Primary spontaneous pneumothorax (PSP) affects 1 in 10 000 people with 10% having affected relatives, so-called familial spontaneous pneumothorax (FSP).1 2 FSPs comprise a diverse group of disorders with extrapulmonary manifestations ranging from renal cancer to arterial dissection.1 2 We previously proposed FSPs be classified into those conditions with aberrant extracellular matrix or defective tumour-suppressor pathways.1 2 A third group of unclassifiable FSPs currently defies diagnosis, but it is unclear if these represent formes frustes of known syndromes or are as yet undescribed disorders.3 Our pneumothorax clinic provided a resource of well-phenotyped individuals who have been assessed by a respiratory-genetics multidisciplinary team (MDT) over a 12-year period. We set out to analyse this cohort, combined with additional whole genome sequencing (WGS) data from the 100 000 Genomes Project, to assess the efficiency of existing clinico-radiological pathways for the diagnosis of FSP.
Methods
Between 2008 and 2020, 492 patients were recruited consecutively from our pneumothorax clinic. Familial cases were reviewed by a pneumothorax genetics MDT comprising respiratory physicians, clinical geneticists, and thoracic radiologists. Detailed methods are provided in online supplemental file 1.
Supplemental material
Results
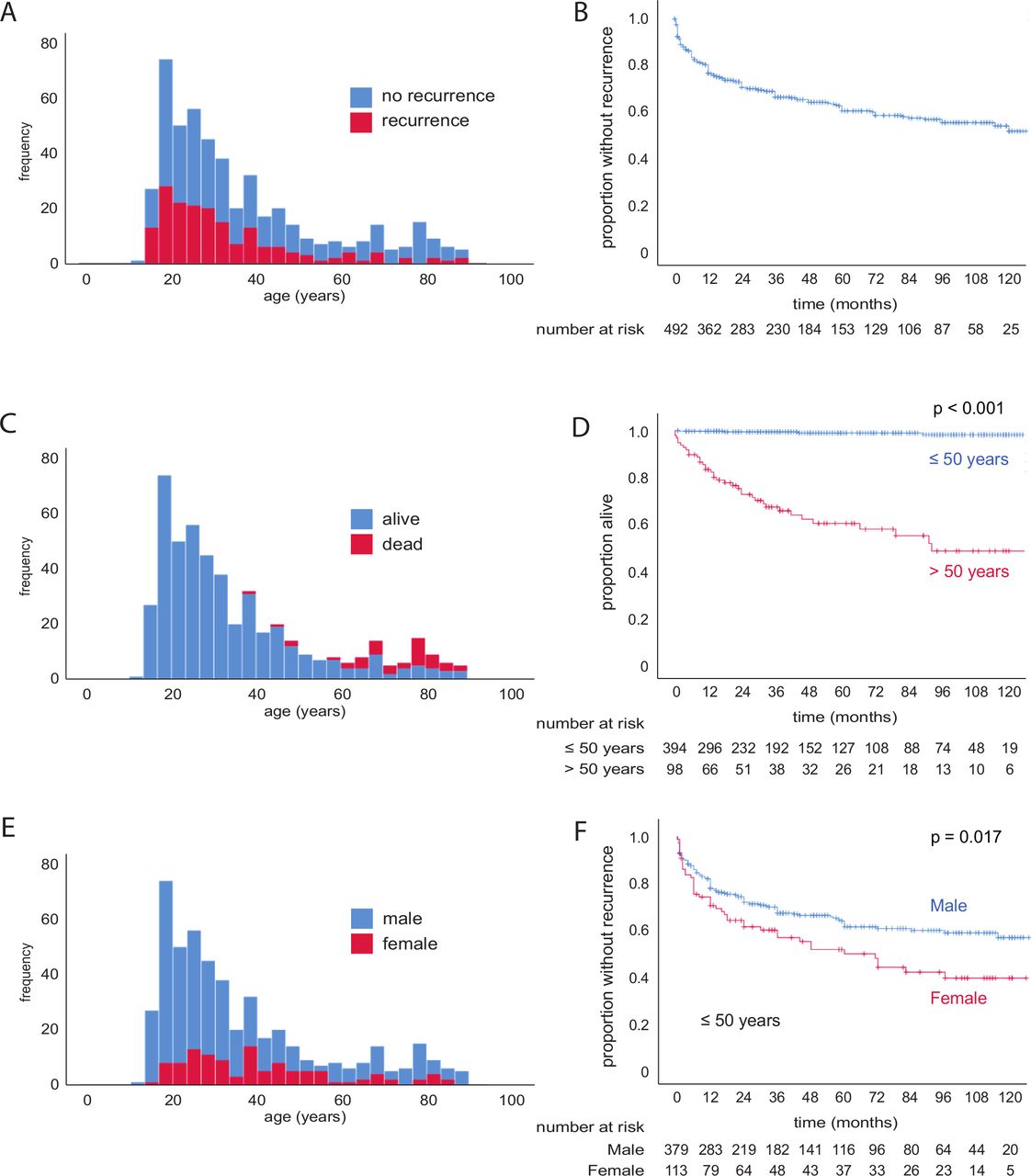
Local demographics biased our cohort towards younger individuals with few cases of secondary spontaneous pneumothorax (SSP) (online supplemental table S1; figure 1A). Overall, 5-year recurrence was 38.6%±2.5%, but this rose to 44.5%±3.4% by 10 years (figure 1B). Five-year recurrence for those 50 years or younger was 41.9%±2.8%, but only 20.7%±4.8% for those >50 years, perhaps owing to higher mortality (39.3%±5.6% at 5 years for >50 years vs 1.0%±0.6% for ≤50 years; figure 1C,D). The comparatively high overall recurrence rate seen in our series may, therefore, reflect the predominance of younger patients. The sex ratio was 3:1 male:female, with no mortality difference between sexes (online supplemental table S1; figure 1E and online supplemental figure S1A). However, 10-year recurrence was significantly higher for females (63.1%±9.4% vs 41.3%±3.5%, p=0.041; online supplemental figure S1B). This persisted when patients >50 years were excluded (p=0.017; figure 1F). Lymphangioleiomyomatosis (LAM) was diagnosed in two women (1.8% of 113 female cases), while thoracic endometriosis (catamenial pneumothorax) was diagnosed in 9 (7.9% of female cases). Both patients with LAM suffered recurrences, as did 7 (77.8%) of the women with catamenial pneumothorax. These conditions may, therefore, contribute to the increased risk of recurrence we observed in women.
Supplemental material

Pneumothorax clinic patent characteristics, survival and pneumothorax recurrence. (A) Histogram of patients; no recurrence (blue), recurrence (red). (B) Kaplan-Meier curve and number at risk table of pneumothorax recurrence. (C) Histogram of patients, alive (blue), died (red). (D) Kaplan-Meier curve and number at risk table of survival; patients 50 years of age or younger (blue), over 50 years old (red). P value calculated by log-rank test. (E) Histogram of patients; male (blue), female (red). (F) Kaplan-Meier curve and number at risk table of pneumothorax recurrence in patients 50 years of age or younger; male (blue), female (red). P value calculated by log-rank test.
A positive family history of pneumothorax was elicited in 14.6% of patients (14.0% of males, 16.8% of females) and was associated with a significantly increased chance of recurrence (figure 2A,B; online supplemental table S1). Overall, 5-year FSP recurrence was 49.6%±6.6% vs 36.7%±2.7% for sporadic pneumothoraces (p=0.034; figure 2A and online supplemental figure S1C). For patients 50 years or younger, 5-year FSP recurrence was 54.7%±7.1% vs 36.2%±2.9% of sporadic pneumothoraces (p=0.025; figure 2B).

{kind=link}
{kind=link}
Familial spontaneous pneumothorax patient characteristics. (A) Histogram of patients; no family history of pneumothorax (blue), familial pneumothorax (red). (B) Kaplan-Meier curve of pneumothorax recurrence in patients 50 years of age or younger; no family history of pneumothorax ‘sporadic’ (blue), familial pneumothorax (red). P value calculated by log-rank test. (C) Relative proportion of monogenic disorder diagnoses made. (D) Thoracic CT scan of patient identified to have Birt-Hogg-Dubé by whole genome sequencing. noted small cyst, one of two that were present. TSC, tuberous sclerosis complex.
Targeted genetic testing guided by a pneumothorax MDT yielded a genetic diagnosis in 26.4% (19/72) of FSP cases (figure 2C). Birt-Hogg-Dubé syndrome was the most common, accounting for 47% of genetic diagnoses (2% of all 492 patients), followed by Marfan (21%), vascular Ehlers-Danlos (11%) and Loeys-Dietz syndromes (11%). Alpha1-antitrypsin deficiency and tuberous sclerosis were more uncommon (5%). We did not observe significant changes in rates of diagnosis over time.
A majority of individuals with FSP (73.6%) failed to fulfil the diagnostic criteria for known genetic disorders. To examine this further, 33 individuals with unclassifiable FSP (27 probands, 6 affected relatives; 64% male) were recruited to the 100 000 Genomes Project4 5 (online supplemental table S2). The crowdsourcing tool PanelApp was used to generate a pneumothorax gene panel. Candidate genes were scored by members of the Genomics England online community and ten genes received sufficient endorsement to be declared ‘green genes’ and included in the pneumothorax panel (table 1). WGS data from the 33 unclassifiable FSP patients were annotated for pathogenicity based on variant segregation within families, frequency in control populations, effect on protein coding, and mode of inheritance. ‘Tier 1’ variants were defined as high-impact variants (eg, likely loss of function) and de novo moderate-impact variants (eg, missense) in this panel. A single tier 1 variant was identified in the genome of one patient: a previously reported pathological variant of FLCN (c.1062+2T>G) indicating a diagnosis of Birt-Hogg-Dubé syndrome. The proband had no cutaneous features of Birt-Hogg-Dubé syndrome and no family history of renal malignancy. The thoracic CT was rereviewed and, in hindsight, noted to show two small cystic lesions (figure 2D). No other tier 1 pathogenic variants were identified indicating that clinico-radiological assessment had been highly successful in excluding known genetic causes of FSP (sensitivity 96%, 26/27 probands).
PanelApp familial pneumothorax gene panel
Discussion
In our cohort of 492 patients with pneumothorax, 14.6% had FSP. Of these, 26.4% were diagnosed clinically with a monogenic disorder, most commonly Birt-Hogg-Dubé syndrome. Previous studies in Chinese and European populations suggested that up to 10% of pneumothoraces are due to FLCN mutations.6 7 In our cohort, the overall proportion was much less at 9 of 492 (2%). To our knowledge, this is the first report of an association between FSP and increased chance of recurrence compared with sporadic pneumothorax. Female sex was also associated with an increased risk of recurrence.
WGS of 33 individuals with unclassifiable FSP revealed a single tier 1 variant indicating that clinico-radiological assessment has high sensitivity for known FSP syndromes, consequently the majority of genetic risk in FSP remains unexplained. A previous genome-wide association study (GWAS) reported an association between PSP and the single nucleotide polymorphism (SNP) rs4733649 in the SLC6A1 gene.8 None of the 33 individuals nor 8854 sequenced non-pneumothorax controls had this SNP so we cannot confirm the association (not shown). COPD is a cause of SSP. Deficiency of α1-antitrypsin is its most common monogenic cause but accounts for under 2% of prevalent cases.9 GWASs have implicated other genes in COPD (HHIP, CHRNA5, HTR4, FAM13A, RIN3, TGFB2, GSTCD-NPNT, CYP2A6 and IL27-CCDC101),10 but no pathogenic variants were identified in these genes in our cases.
An important limitation of our study is its geographical restriction to one centre. National analysis of FSP will be necessary to determine its population prevalence.
In summary, assessment by a pneumothorax MDT has high sensitivity for diagnosis of monogenic causes of FSP by enabling targeted genetic testing. We find FSP increases the risk of recurrence compared with sporadic pneumothoraces. A substantial proportion of the genetic risk for FSP remains unaccounted for, but ongoing analysis of the 100 000 Genomes dataset will address this.
Ethics statements
Ethics approval
The study was approved by the University of Cambridge Human Biology Research Ethics Committee HBREC.2020.28.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @MarciniakLab
Contributors SJM is the guarantor of the content of the manuscript. HLG, JB, ERM and SJM were involved in the conception and design, data analysis and interpretation. HLG performed the data collection. All authors were involved in drafting the submitted article.
Funding This work was supported by NIHR Cambridge Biomedical Campus grant number BRC-1215-20014.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.