Article Text

Abstract
Background Although a variety of pathological changes have been described in small airways of patients with COPD, the critical anatomic features determining airflow limitation remain incompletely characterised.
Methods We examined lung tissue specimens from 18 non-smokers without chronic lung disease and 55 former smokers with COPD for pathological features of small airways that could contribute to airflow limitation. Morphometric evaluation was performed for epithelial and subepithelial tissue thickness, collagen and elastin content, luminal mucus and radial alveolar attachments. Immune/inflammatory cells were enumerated in airway walls. Quantitative emphysema scoring was performed on chest CT scans.
Results Small airways from patients with COPD showed thickening of epithelial and subepithelial tissue, mucus plugging and reduced collagen density in the airway wall (in severe COPD). In patients with COPD, we also observed a striking loss of alveolar attachments, which are connective tissue septa that insert radially into the small airway adventitia. While each of these parameters correlated with reduced airflow (FEV1), multivariable regression analysis indicated that loss of alveolar attachments was the major determinant of airflow limitation related to small airways. Neutrophilic infiltration of airway walls and collagen degradation in airway adventitia correlated with loss of alveolar attachments. In addition, quantitative analysis of CT scans identified an association between the extent of emphysema and loss of alveolar attachments.
Conclusion In COPD, loss of radial alveolar attachments in small airways is the pathological feature most closely related to airflow limitation. Destruction of alveolar attachments may be mediated by neutrophilic inflammation.
- COPD pathology
- emphysema
Data availability statement
Research data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is the key question?
This study addresses the question of which pathological features of small airways in COPD are most important in determining airflow limitation.
What is the bottom line?
Destruction of radial alveolar attachments between small airways and alveolar tissue, which is associated with neutrophilic inflammation in airway walls, is a major factor responsible for lung function decline in COPD.
Why read on?
Understanding the critical features of small airway pathology in COPD is essential for designing new therapeutic interventions to preserve or improve lung function.
Introduction
COPD is a complex and heterogeneous clinical syndrome characterised by fixed airflow limitation.1–3 While airflow limitation in COPD has been attributed to pathology in both conducting airways and alveolar tissue, seminal work by James Hogg’s group showed that small airways (<2 mm in internal diameter) are the primary site for airflow limitation in this disease.4 Additional studies have shown that a reduction in the number of small airways on microCT scanning occurs prior to the onset of emphysematous destruction,5 suggesting that small airways may be the site where initial pathological changes develop in COPD.
Understanding the pathological features that lead to functional alterations in COPD is crucial for developing treatments that limit disease progression or restore function. While a number of histopathologic abnormalities in small airways have been reported in COPD, the relative importance of each of these features in determining airflow limitation in COPD remains unclear. To investigate the physiological impact of individual pathological changes in small airways of patients with COPD, we used quantitative histochemistry to examine all components of the airway wall, including inner (epithelial thickness, mucus plugging), mural (wall thickness, collagen and elastin density) and outer (number of alveolar attachments) compartments. We then evaluated the relative importance of each parameter in determining airflow limitation. In addition, we assessed associations between these parameters and: (1) accumulation of immune/inflammatory cells in airway walls and (2) the extent of emphysema on CT scans.
Methods
Specimens and patient population
Severity of airflow limitation in COPD was determined using the Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria.6 7 Lung tissue specimens were obtained from eight lifelong non-smokers (NSs), 10 former smokers (FSs) without chronic lung disease, and 31 FSs with mild, moderate or severe COPD (GOLD stages I–III) at the time of lung cancer resection. Airways from tissue outside the tumour margins were analysed for these studies. Tissue specimens from an additional 24 patients with very severe COPD (GOLD stage IV) were obtained from explanted lungs at the time of lung transplantation. Table 1 summarises the demographic and clinical characteristics of study subjects. Lung specimens were originally collected with approval from the Institutional Review Board at Vanderbilt University Medical Center (protocol #s 060162 and 000616). This study was performed with deidentified specimens and was approved by the Institutional Review Board at Vanderbilt University Medical Center (protocol #161046).
Clinical characteristics of study participants
Airway histology and morphometry
All airways meeting morphological criteria for ‘bronchiole’ or ‘small conducting airway’ were examined. Airways containing mural cartilage or submucosal glands as well as airways >2 mm in internal diameter were considered large airways and excluded from analysis. Smaller respiratory bronchioles with discontinuous walls that opened into alveolar ducts were also excluded. Table 1 shows the number of airways examined, along with the basement membrane length, for each group.
Paraffin sections (5 µm) were cut from each tissue block (two tissue blocks for each study participant) and serial sections were stained with H&E for routine histologic evaluation, periodic acid-Schiff staining for detection of mucin or PicroSirius red-staining for analysis of collagen density. Additional serial sections were used for immunostaining to detect immune inflammatory cells (CD4 or CD8 T cells, CD19 B cells or neutrophils), structural cells (smooth muscle) and elastin. The list of antibodies and immunohistochemistry protocols are available in the online supplemental file.
Supplemental material
Alveolar attachments were identified and counted for each small airway and normalised to reticular basement membrane (RBM) length. The length of basement membrane contiguous with the adjacent pulmonary vessel, as well as attachments to vascular adventitia, were excluded from the measurements.
To assess airway wall remodelling, epithelial area (the difference between the area delineated by the subepithelial RBM and the apical epithelial surface) and subepithelial area (the difference between the area delineated by the RBM and the outer edge of adventitia) were measured for each airway on digital microscopic images (×10 objective). These measurements are presented as a ratio of volume to surface area (V:SAEPI or V:SASubEPI) since they represent volume and surface area in three dimensions, as described by Hogg et al.8 The relationship between epithelial height and intraluminal mucus and luminal area of each airway was performed as described in online supplemental figure 1.8 9 Density for collagen and elastin was assessed on PicroSirius red-stained or immunostained sections, respectively, by detection of red fluorescence. Each airway was captured with the same magnification (×10 objective) and the same digital camera settings. Density was calculated as the area containing a specific fluorescence/immunofluorescence signal within the total area of the airway subepithelium. Separately, collagen and elastin density measured in the outer 10 µm zone of adventitia.
Neutrophils, CD4 or CD8 T cells were enumerated within the airway wall between the epithelial surface and outer edge of adventitia and normalised to the length of RBM (per mm) or area of the airway wall (mm2). The percentage of airways with adjacent lymphoid follicles was assessed on sections immunostained for CD19-positive B cells.
All morphometric measurements were performed using Image-Pro Plus V.7.0 software (Media Cybernetics, Silver Springs, Maryland, USA) or ImageJ V.1.8.0 (NIH, Bethesda, Maryland, USA). Since the lungs examined in this study were not inflated prior to fixation, morphometric measurements were normalised to airway RBM length according to ATS/ERS standards for Quantitative Assessment of Lung Structure.10
CT-based classification of emphysema
Quantitative emphysema scoring was performed using computational software provided by Ziosoft USA Inc (Belmont, California, USA) from reconstructed coronal images obtained from chest CT scans.11 Emphysema scoring was calculated utilising low attenuation volume analysis on the most recent CT scan prior to lung resection or transplantation.
Statistical analysis
We conducted pairwise comparisons among study groups (NS, FS, COPD I–II and COPD III–IV) using Student’s t-test or Wilcoxon rank-sum test. Estimated p values were Bonferroni-adjusted. Pearson’s correlation analyses and linear regressions were performed for each pathological parameter with FEV1 and each inflammatory parameter with alveolar attachment counts. We applied a multivariable linear regression model to examine the relationships between FEV1 and measured pathological parameters. Based on variance decomposition by the LMG (Lindeman, Merenda, Gold) method, we assessed the relative contribution of each parameter on FEV1 using the proportion of FEV1 variability explained by assessed parameters.12 13 In addition, we performed the same analysis excluding patients with COPD with severe disease (GOLD stages III–IV). A similar approach was used to assess the contribution of inflammatory parameters to loss of alveolar attachments. We performed a model-based causal mediation analysis to assess the relationship between neutrophils, alveolar attachments and FEV1 using the non-parametric bootstrap method of simulation (number of simulations equal 1000). Percentile intervals were calculated for bootstrap-based confidence intervals.14 15 Both models (mediator and outcome) were specified based on relative importance analyses and were linear. The average indirect effect, direct effect and total effect were assessed. Normality of residuals of linear models was assessed and the normal distribution assumption was met. A p value <0.05 was considered statistically significant. All analyses were performed using R-software V.3.5.2 (www.r-project.org).
Results
Small airway pathology in COPD
Our approach was to quantitatively relate pathologic features of small airways to airflow (FEV1) in individual patients with COPD and control subjects. We determined the pathology present in the inner/epithelial, mural and outer/adventitial compartments of small airways from patients with COPD compared with lifelong NS and FS without COPD.
Inner
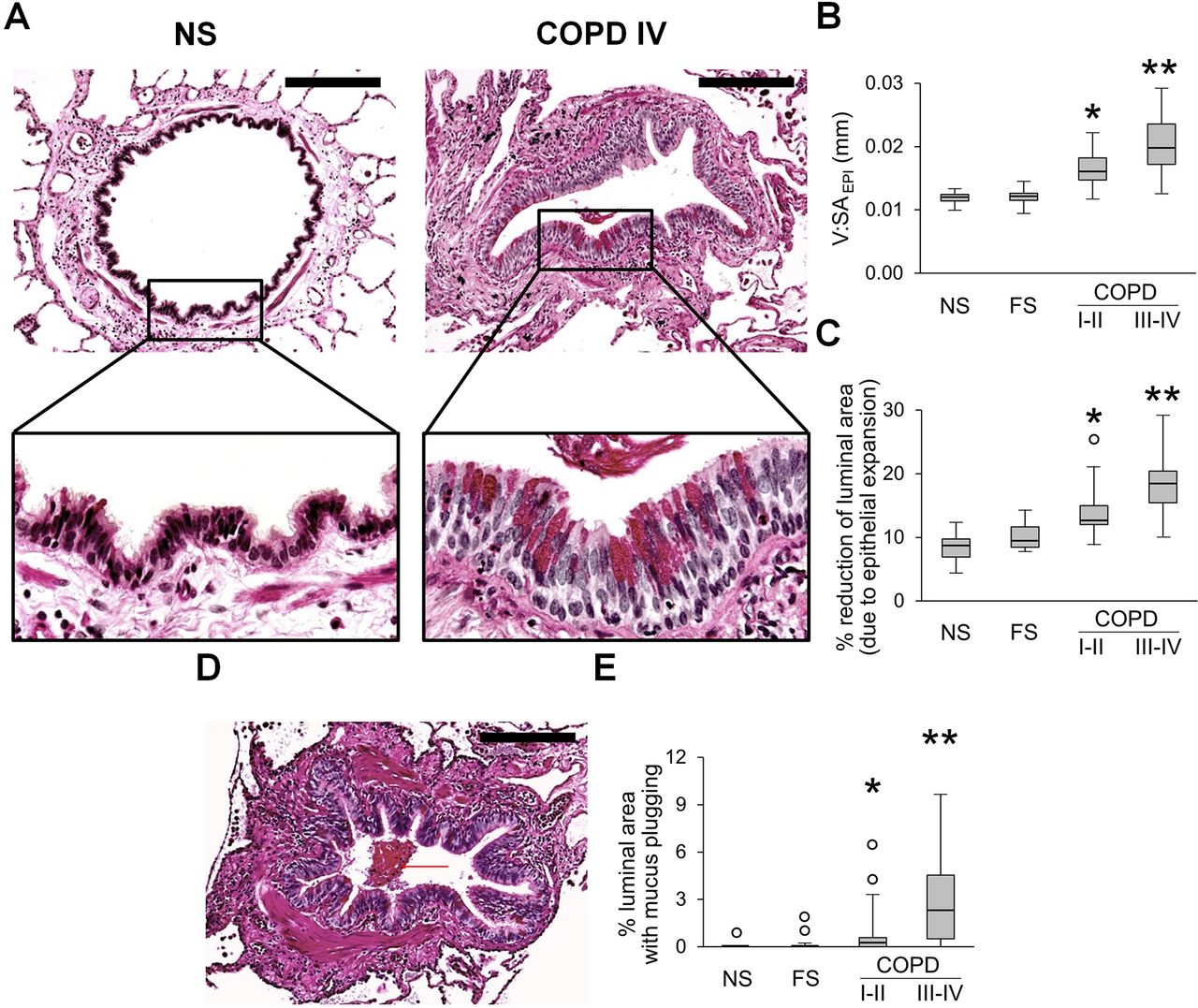
Evaluation of the epithelial compartment in small airways revealed that the epithelial layer had widespread goblet cell metaplasia and increased epithelial height in patients with COPD, thus reducing the cross-sectional area of the airway lumen (figure 1A–C). In addition, we observed a progressive accumulation of mucus plugs within airways from GOLD stage I–II to GOLD stage III–IV COPD, also reducing luminal area (figure 1D,E).
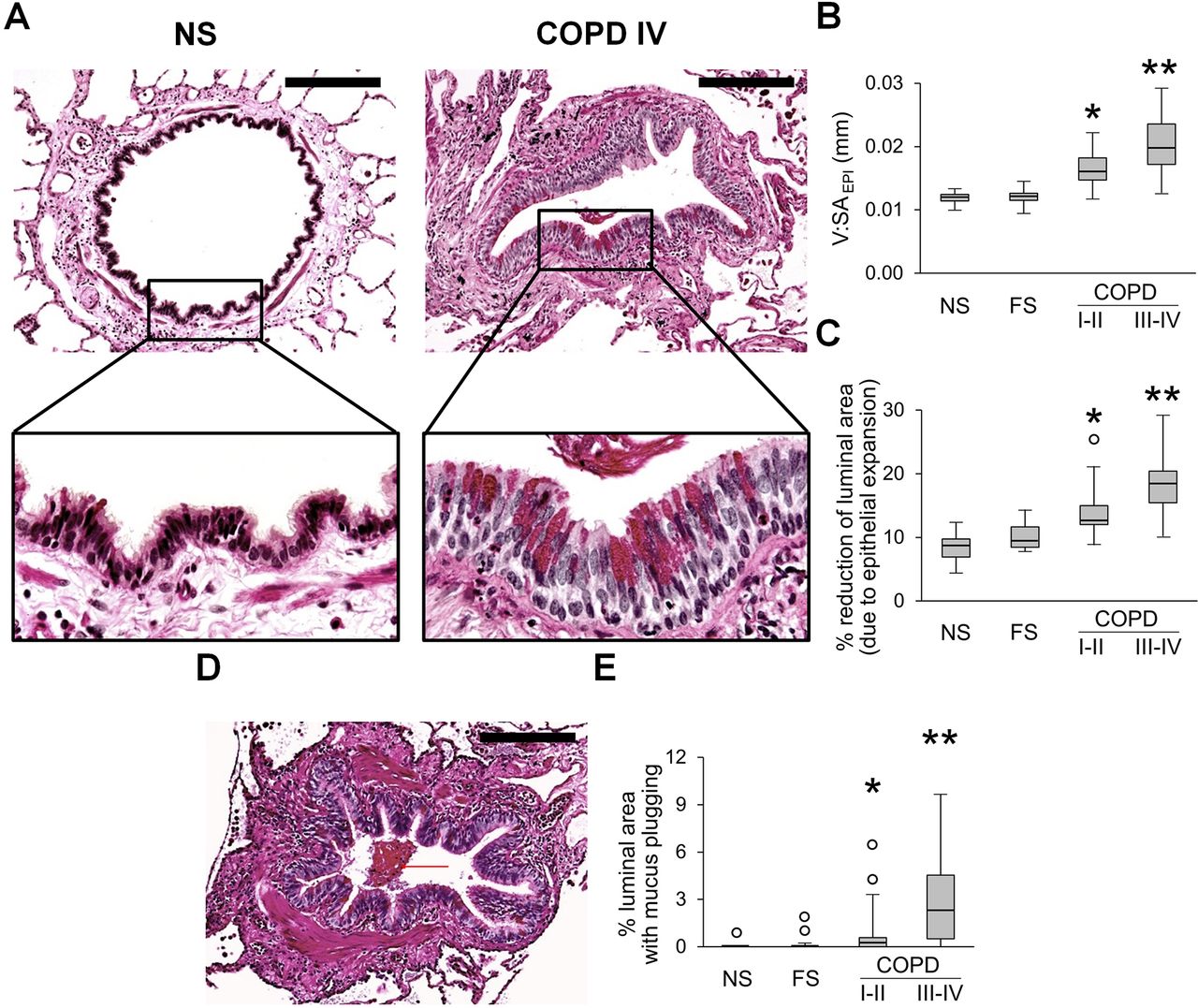
Epithelial remodelling and mucus accumulation in small airways of patients with COPD. (A) Representative images of normal-appearing structural organisation of small airway in lifelong NS subject and a combination of goblet cell metaplasia and increased epithelial height in small airway from a patient in GOLD stage IV COPD. PAS staining. Scale bars=100 µm. Inserts show detailed characteristics of epithelial remodelling. (B, C) Box and whisker plots showing progressive increase of epithelial height (V:SAepi) (B) and reduction of airway luminal area due to epithelial expansion (C) in COPD small airways compared with NS and FS airways. (D) Mucus plug in the airway lumen. PAS staining. Scale bar=100 µm. (E) Box and whisker plots showing progressive reduction of airway luminal area due to mucus plugging in COPD small airways compared with NS and FS airways. Boxes represent the IQR, whiskers extend to the most extreme data point which is no more than 1.5 times the IQR from the box, and circles beyond the whiskers are extreme values, the line within the box represents the median. Groups were compared pairwise using Mann-Whitney U test. Estimated p values were Bonferroni-adjusted. The threshold for significance was 0.05. *Significantly different compared with NS controls, **significantly different compared with all other groups. FS, former smoker; GOLD, Global Initiative for Chronic Obstructive Lung Disease; NS, non-smoker; PAS, periodic acid-Schiff.
Mural
We evaluated the mural compartment of the airway wall by measuring the thickness of the subepithelial compartment and quantifying collagen and elastin content in the extracellular matrix (ECM) (figure 2). We observed enlargement (thickening) of the airway subepithelium in COPD airways compared with NS and FS controls (figure 2A,B). Because ECM is a key component of the airway wall and alterations in connective tissue are believed to be crucial pathophysiological processes, we next analysed collagen by PicroSirius red-staining and found increased collagen density in patients with GOLD stage I–II COPD, as well as FS controls, compared with NS airways. In contrast, small airways of patients with GOLD stage III–IV COPD were characterised by decreased collagen density compared with all other groups (figure 2C). Despite the overall reduction in collagen content in airways from patients with severe COPD, areas of collagen deposition were detected in the subepithelial RBM and in perivascular areas (figure 2A, yellow arrows), representing a combination of localised fibrous remodelling along with a diffuse destructive process. As measured by immunofluorescence, elastin density was similar in the airway subepithelium in all groups, suggesting that reduced elastin content is not a major feature of airway remodelling in COPD (figure 2D).
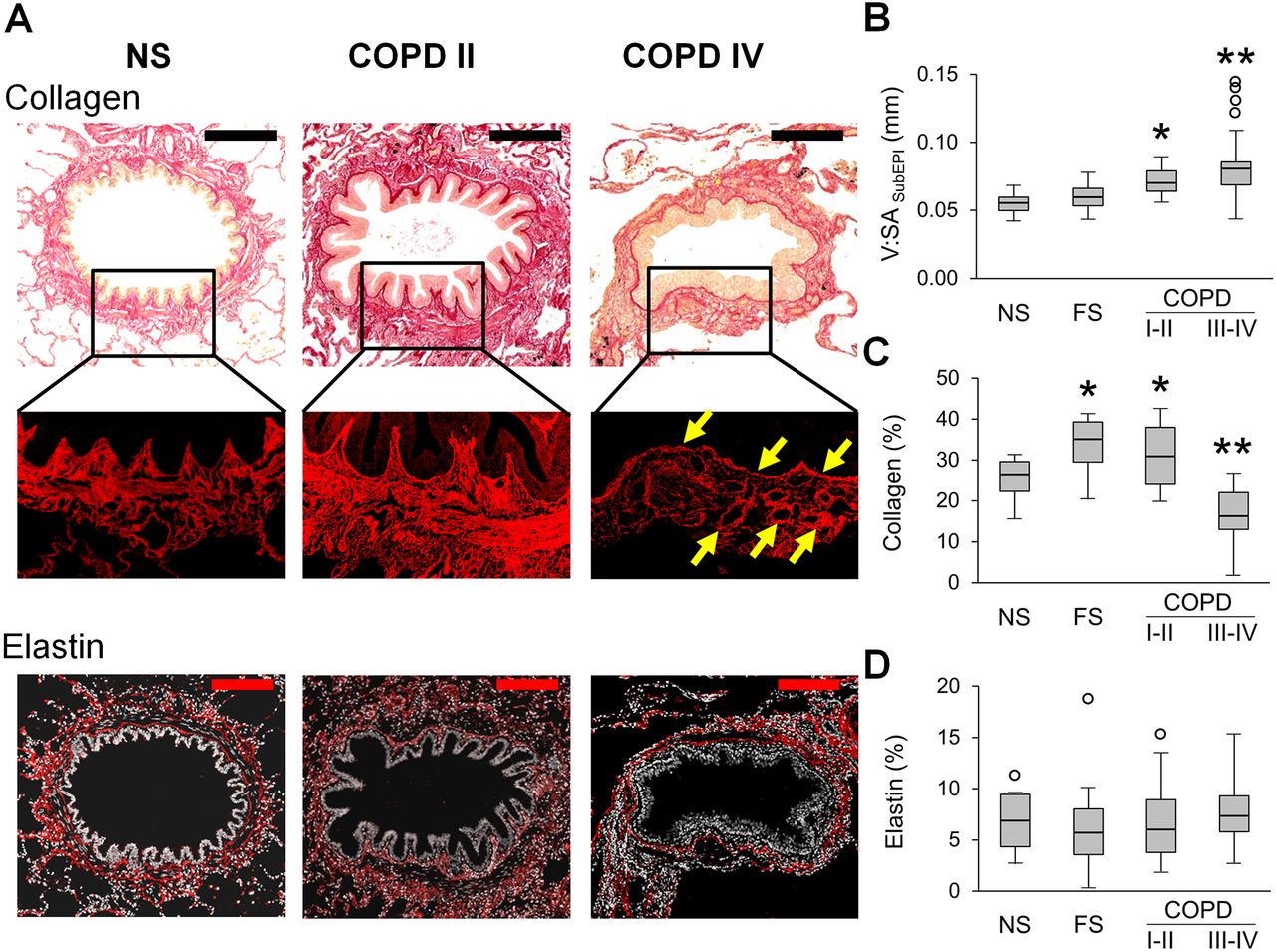
Mural remodelling of small airways of patients with COPD. (A) Representative images of normal-appearing structural organisation of a small airway wall in lifelong NS subject and wall remodelling in patients with GOLD stage II COPD and stage IV COPD. PicroSirius red-staining. Scale bar=100 µm. Inserts demonstrate PicroSirius red-staining (P-red) under red fluorescence. Fine structural organisation of collagen fibres in NS airway, collagen deposition in small airway from a patient in GOLD stage II COPD, and a combination of collagen deposition in RBM and perivascular regions (yellow arrows) with collagen scaffold degradation in small airway from a patient in GOLD stage IV COPD. Bottom images show elastin immunostaining in small airways from lifelong NS subject, patients with GOLD stage II and stage IV COPD. Scale bars=100 µm. (B–D) Box and whisker plots show wall thickness (V:SAsubepi) (B), collagen density (C) and elastin density (D) in small airways of NS and FS controls and patients with COPD. Boxes represent the IQR, whiskers extend to the most extreme data point which is no more than 1.5 times the IQR from the box and circles beyond the whiskers are extreme values, the line within the box represents the median. Groups were compared pairwise using Student’s t-test for collagen content and Mann-Whitney U test otherwise. Estimated p values were Bonferroni-adjusted. The threshold for significance was 0.05. *Significantly different compared with NS controls, **significantly different compared with all other groups. FS, former smoker; GOLD, Global Initiative for Chronic Obstructive Lung Disease; NS, non-smoker.
Outer
Small airways are connected to the surrounding parenchyma by radial attachments that emanate from interalveolar septa and insert into the airway adventitia (figure 3A). We counted the number of alveolar attachments in each small airway appearing on cross-section and normalised to RBM length. This measurement showed a modest reduction of alveolar attachments in GOLD stage I–II COPD and a dramatic loss of attachments (>50%) in GOLD stage III–IV COPD compared with control groups (figure 3B).

Reduced alveolar attachments around small airways of patients with COPD. (A) Representative images of intact alveolar attachments in small airway from lifelong NS subject and their loss in small airway from a patient in GOLD stage IV COPD. PAS staining. Scale bars=100 µm. Inserts show detailed organisation of alveolar attachments (red stars). (B) Box and whisker plots showing progressive loss of alveolar attachments in COPD small airways compared with NS and FS airways. Boxes represent the IQR, whiskers extend to the most extreme data point which is no more than 1.5 times the IQR from the box, and circles beyond the whiskers are extreme values, the line within the box represents the median. Groups were compared pairwise using Student’s t-test. Estimated p values were Bonferroni-adjusted. The threshold for significance was 0.05. *Significantly different compared with NS controls, **significantly different compared with all other groups. FS, former smoker; GOLD, Global Initiative for Chronic Obstructive Lung Disease; NS, non-smoker; PAS, periodic acid-Schiff.
Together, these data show that all compartments of the airway wall (inner, mural, outer) are involved in the complex pathology of COPD.
Loss of alveolar attachments is the major determinant of airflow limitation and correlates with adventitial collagen degradation
Having quantified pathological changes in small airways of patients with COPD, we next analysed the association of each pathological feature of small airway disease with airflow (FEV1) measured on the most recent pulmonary function testing prior to lung resection or explant (figure 4A–F). Several pathologic features correlated with FEV1; however, alveolar attachment counts had the most striking correlation (r=0.870). Multivariable linear regression analysis followed by LMG variance decomposition showed that 80.3% of the variance in FEV1 could be attributed to small airway pathology and confirmed that loss of alveolar attachments was the major small airway determinant of airflow limitation (figure 4G). Since small airway pathological features vary in mild/moderate and advanced stages of disease, we performed the same analysis excluding patients with severe COPD (GOLD stages III–IV). Similar to the entire cohort, loss of alveolar attachments was identified as a major contributor to airflow limitation in patients with mild-to-moderate COPD (GOLD stages I–II). Other parameters, particularly increased epithelial height, also appeared to play a role (online supplemental figure 2).
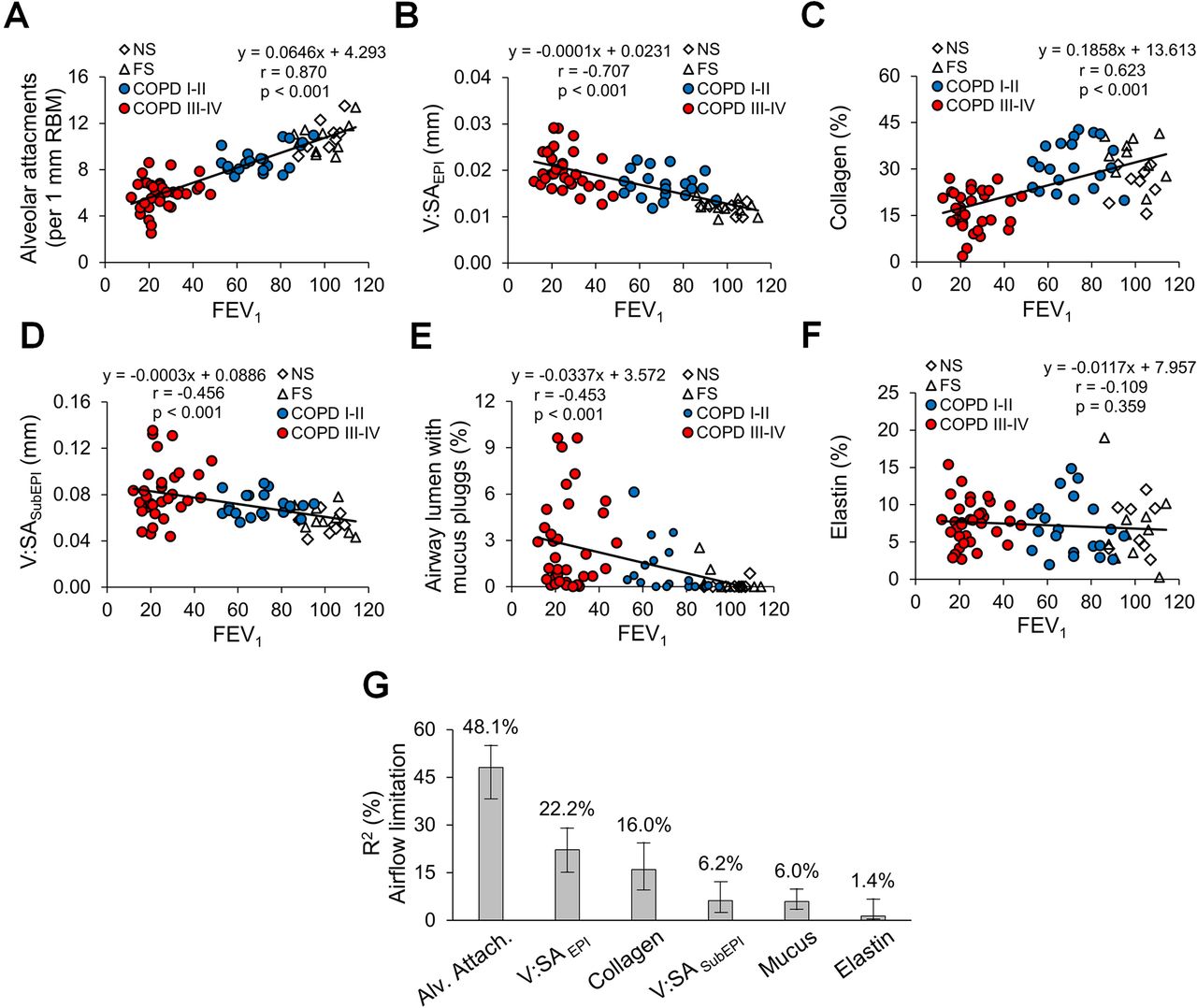
Relative contribution of small airway pathological findings to airflow limitation in COPD. (A–E) Scatter plots showing correlations and lines of best fit of alveolar attachments (A), epithelial height (B), mucus plugging (C), airway wall thickness (D), collagen density (E) and elastin density (F) with airflow limitation measured as FEV1. (G) Relative contribution of pathological parameters on airflow limitation (measured as FEV1) plotted as the proportion of FEV1 variability and its corresponding 95% CI explained by each parameter. R²=80.3%, metrics are normalised to sum 100%. FS, former smoker; GOLD, Global Initiative for Chronic Obstructive Lung Disease; NS, non-smoker.
Since the connection between small airways and surrounding alveolar tissue requires a continuous extra cellular matrix, we sought to better define the pathology of alveolar attachments. Specifically, we performed histologic analysis of anchoring collagen and elastin fibres at the site of insertion of alveolar attachments within the small airway adventitia. In contrast to NS and FS controls, COPD airways showed fragmentation of collagen and elastin anchoring fibres, particularly in severe COPD, consistent with a localised destructive process (figure 5A). Assessment of collagen fibres in the outer adventitia (10 µm zone) revealed a significant reduction of collagen density in patients with GOLD stage III–IV COPD (figure 5B). Although total elastin density in airway adventitia remained unchanged in COPD airways compared with NS and FS airways (figure 5C), fragmentation of elastin fibres was apparent at insertion sites for alveolar attachments in COPD airways (figure 5A). Correlation and linear regression analyses between the number of alveolar attachments and adventitial collagen density (figure 5D) suggested collagen degradation could be a precursor of attachment loss.
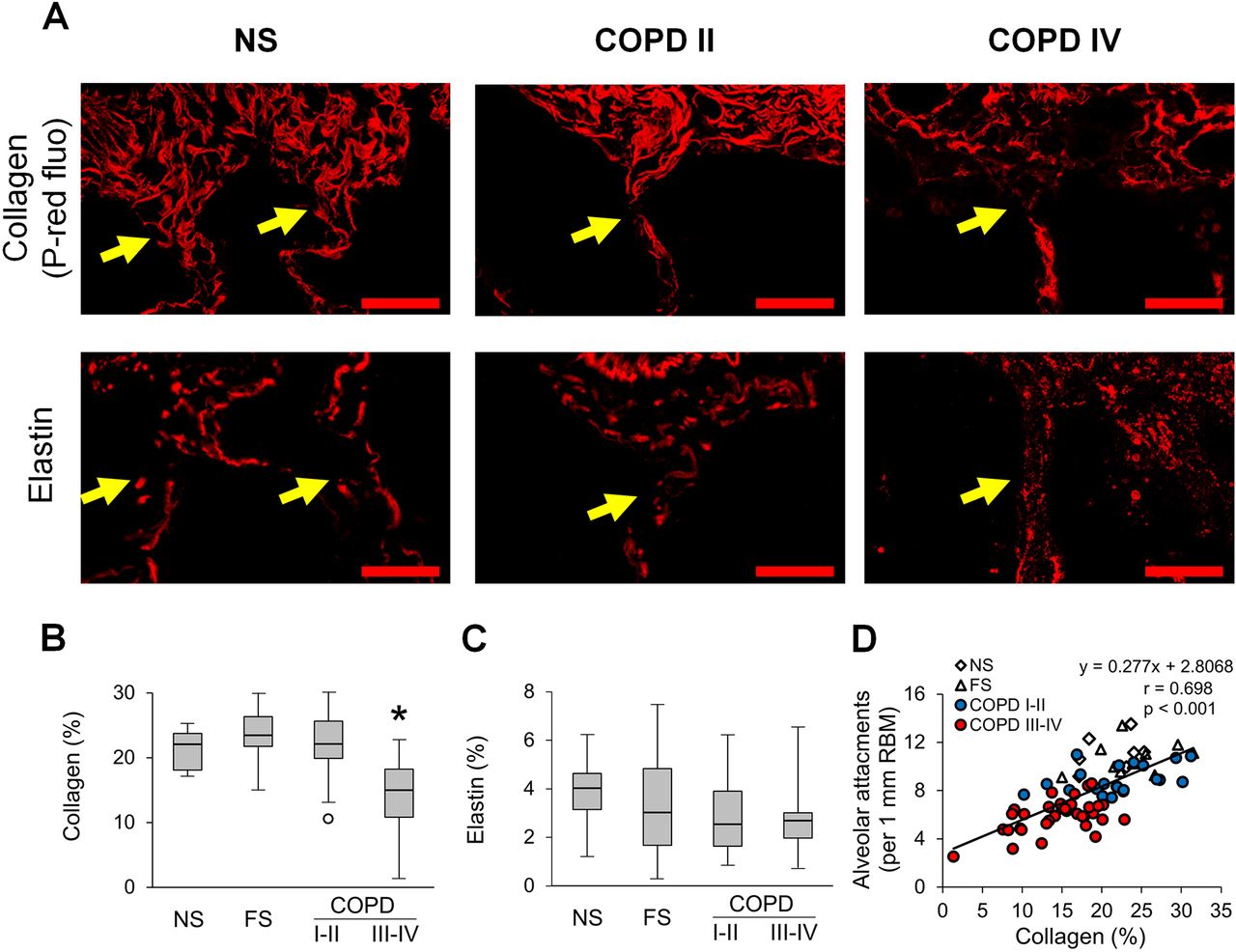
Collagen degradation in the adventitia of small airways is associated with loss of alveolar attachments. (A) Representative images of collagen and elastin anchoring fibres (yellow arrows) in alveolar attachments in a lifelong NS subject, patient with GOLD stage II COPD, and a patient with GOLD stage IV COPD. Top row: PicroSirius red-staining under red fluorescence; bottom row: elastin immunofluorescence. Scale bars=20 µm. (B,C) Box and whisker plots show collagen (B) and elastin (C) density in 10 µm zone of outer adventitia in small airways from NS and FS controls and patients with COPD. Boxes represent the IQR, whiskers extend to the most extreme data point which is no more than 1.5 times the IQR from the box, and circles beyond the whiskers are extreme values, the line within the box represents the median. Groups were compared pairwise using Mann-Whitney U test. Estimated p values were Bonferroni-adjusted. The threshold for significance was 0.05. *Significantly different compared with all other groups. (D) Scatter plot showing correlation and best fit line of collagen density in outer adventitia of small airways with alveolar attachments. FS, former smoker; GOLD, Global Initiative for Chronic Obstructive Lung Disease; NS, non-smoker.
Inflammation relates to airflow limitation and loss of alveolar attachments
Since inflammation plays a key role in COPD pathogenesis and is believed to directly lead to connective tissue destruction, we investigated the relationship of airway inflammation to airflow limitation and alveolar attachment loss. To accomplish this, we quantified lymphocytes and neutrophils in airway walls. Analysis of CD4 and CD8 T lymphocytes revealed increased numbers of these cells in airway walls of patients with COPD when normalised to RBM length (figure 6A,B). However, when CD4 and CD8 T cell influx was normalised to airway wall area, only CD8 T cells showed a modest increase in patients with COPD (online supplemental figure 3A,B). B cell-containing bronchus-associated lymphoid follicles were detected primarily in GOLD stage III–IV COPD (figure 6C). In contrast to lymphocytes, neutrophils were progressively increased in small airways from GOLD stage I–II COPD to GOLD stage III–IV COPD when normalised for either RBM length or airway wall area (figure 7A,B, online supplemental figure 3C).
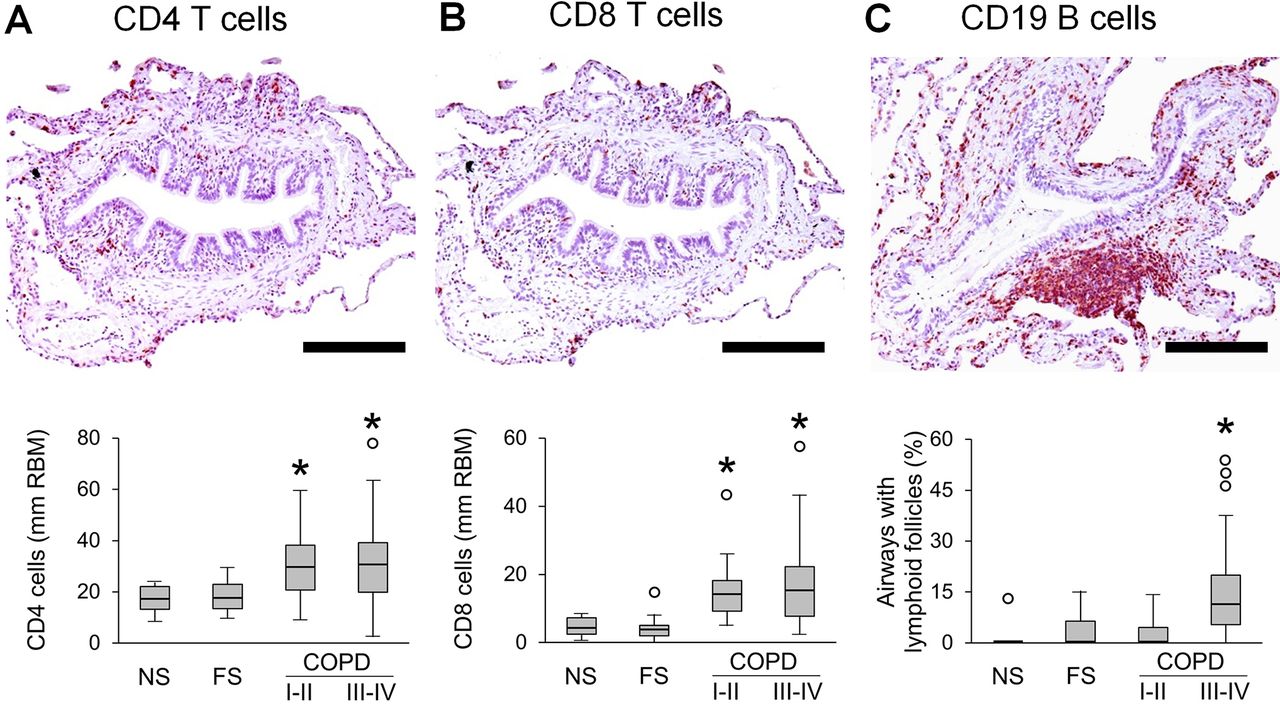
Quantification of lymphocytes in small airways of patients with COPD. (A–C) Representative images of CD4 (A) and CD8 (B) T lymphocyte infiltration in airway wall and lymphoid follicle adjacent to small airway (C). Box and whisker plots show lymphocyte counts in airway walls with normalisation to 1 mm RBM, and the proportion of small airways with adjacent lymphoid follicles from all patient groups. Boxes represent the IQR, whiskers extend to the most extreme data point which is no more than 1.5 times the IQR from the box and circles beyond the whiskers are extreme values, the line within the box represents the median. Groups were compared pairwise using Mann-Whitney U test. Estimated p values were Bonferroni-adjusted. The threshold for significance was 0.05. *Significantly different compared with NS controls. FS, former smoker; NS, non-smoker; RBM, reticular basement membrane.

Quantification of neutrophils in small airways of patients with COPD. (A) Representative image of neutrophil infiltration in airway wall. (B) Box and whisker plots show neutrophil counts in airway walls with normalisation to 1 mm RBM. Boxes represent the IQR, whiskers extend to the most extreme data point which is no more than 1.5 times the IQR from the box, and circles beyond the whiskers are extreme values, the line within the box represents the median. Groups were compared pairwise using Mann-Whitney U test. Estimated p values were Bonferroni-adjusted. The threshold for significance was 0.05. *Significantly different compared with NS controls, **significantly different compared with all other groups. (C) Scatter plot showing correlation and best fit line between small airway wall neutrophilic infiltration and FEV1. (D) Scatter plot showing correlation and best fit line between small airway wall neutrophilic infiltration and alveolar attachment counts. (E) Multivariable regression analysis with following LMG variance decomposition showing the relative contribution and importance of inflammatory parameters on alveolar attachment loss plotted as the proportion of variability and its corresponding 95% CI explained by each parameter. Multivariable regression model R²=43.9%, relative importance metrics are normalised to 100%. FS, former smoker; LMG, Lindeman, Merenda, Gold; NS, non-smoker; RBM, reticular basement membrane.
FEV1 showed a negative correlation (r=−0.664) with neutrophil counts in airway walls (figure 7C). In addition, we found significant inverse associations between FEV1 and CD4 T cells or CD8 T cells in airway walls (normalised for RBM length), as well as B cell containing lymphoid follicles adjacent to small airways (online supplemental figure 4A–C).
Next, we analysed correlations between immune/inflammatory cells and alveolar attachments, and found that neutrophils, CD8 T cells and lymphoid follicles were associated with attachment loss (Figure 7D and online supplemental figures 4D–F). Multivariable linear regression analysis showed that 43.9% of the variance in alveolar attachment number could be explained by inflammatory cell infiltration into airway walls, with neutrophils being the most important contributor according to LMG variance decomposition (figure 7E). Finally, mediation analysis indicated that 49% of the effects of neutrophilic inflammation on FEV1 could be attributed to loss/destruction of alveolar attachment (p<0.001).
Radiographic emphysema correlates with loss of alveolar attachments in small airways
Alveolar attachments arise from the interalveolar septa and their loss represents destruction of a specific portion of alveolar wall. Since destruction of alveolar walls is the defining feature of emphysema, we determined whether loss of alveolar attachments in COPD airways was associated with emphysema as measured on chest CT scans. As expected, quantification of low attenuation areas on CT scans revealed low emphysema scores in the lungs of NS and FS controls and patients with GOLD stage I–II COPD; however, emphysema scores were markedly increased (although highly variable) in lungs of patients with GOLD stage III–IV COPD (figure 8A,B). The percentage of emphysema (low attenuation) showed a negative correlation with airflow as measured by FEV1 (figure 8C). In addition, we identified an inverse relationship between alveolar attachments and degree of radiographic emphysema (figure 8D), suggesting a possible connection between the destructive processes of diffuse emphysema and localised destruction of attachments around diseased small airways.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Relationship of emphysema to loss of alveolar attachments in COPD. (A) Coronal CT images for patient with very severe COPD (GOLD stage IV). Areas of emphysema (low attenuation) are coloured blue. (B) Box and whisker plots showing per cent of lungs containing LAV as an indicator of emphysema in different patient groups. Boxes represent the IQR, whiskers extend to the most extreme data point which is no more than 1.5 times the IQR from the box and circles beyond the whiskers are extreme values, the line within the box represents the median. Groups were compared pairwise using Mann-Whitney U test. Estimated p values were Bonferroni-adjusted. The threshold for significance was 0.05. *Significantly different compared with all other groups. (C) Scatter plot showing correlation and best fit line between percentage of lung with emphysema and FEV1. (D) Scatter plot showing correlation and best fit line between percentage of lung with emphysema and alveolar attachment counts. FS, former smoker; GOLD, Global Initiative for Chronic Obstructive Lung Disease; LAV, low attenuation volume; NS, non-smoker; PAS, periodic acid-Schiff.
Discussion
Our study provides important information about the structural basis of airflow limitation in COPD and bolsters the concept that small airway pathology is a central determinant of disease development and progression. Among the pathological changes linked to small airway dysfunction in COPD, we found that loss of radial alveolar attachments is the best correlate of airflow limitation. This finding suggests that destructive processes in the airway wall, rather than fibrotic thickening, plays a central role in lung function decline during COPD progression. Further, the close correlation between neutrophil invasion into the airway wall and loss of alveolar attachments suggests that these cells may mediate attachment destruction. Finally, loss of collagen in the region of the alveolar attachments suggests that destruction of collagenous ECM may be a key component of this process.
Our data support the proposal recently espoused by Janssen and Wouters16 that loss of alveolar attachments is the most plausible link between small airways disease and emphysema. While it has been suggested that loss of alveolar attachments occurs secondary to emphysematous destruction of lung parenchyma,17–19 our data support the possibility that airway inflammation, rather than inflammation in lung parenchyma, is the driving force for destructive processes in the outer zone of airway adventitia that lead to attachment loss. This idea is supported by the tight correlation between airway wall neutrophils and loss of alveolar attachments, as well as the finding that loss of alveolar attachments is more closely associated with collagen destruction in the airway adventitia than with the degree of radiographic emphysema. This ‘inside-out’ paradigm for inflammation and tissue destruction originating in small airways in COPD, previously described by Mitzner,20 could explain both the initiation of the disease in small airways and centrilobular progression of emphysema from the centre of secondary pulmonary lobules to surrounding interlobular septa.
Alveolar attachments are composed of connective tissue septa that insert radially into the adventitia of small airways.21 Loss of alveolar attachments is thought to untether small airways from adjacent alveolar tissue, thereby contributing to dynamic airflow limitation through reduced radial tension and premature closure of small airways during expiration.21 22 Reduced numbers of alveolar attachments in small airways was first described in 1962 in conjunction with parenchymal emphysema.23 Further data supporting an association between alveolar attachment loss and localised emphysema comes from a recent study using parametric response mapping of CT images to differentiate regions of the lungs with ‘functional small airways disease’ versus emphysema.24 These investigators found that alveolar attachment loss occurred predominantly in regions of the lung with emphysema. In contrast, other studies have raised the possibility that destruction of alveolar attachments may occur separately from parenchymal emphysema by demonstrating loss of alveolar attachments in current smokers and patients with early stage COPD who do not have substantial emphysema.17–19 25
Although several immune/inflammatory cell types (including neutrophils and lymphocytes) are reported to be involved in COPD pathology,26 27 we found a close correlation between neutrophil invasion into the airway walls and alveolar attachment loss, implicating these cells as the culprit for destruction of alveolar attachments. These findings are consistent with our prior studies showing that neutrophil infiltration of COPD small airways is related to localised structural epithelial abnormalities and breakdown of the mucosal immune barrier.28 29 We have shown that localised deficiency of secretory IgA (SIgA) on the airway surface, related to reduced expression of the transport protein polyimmunoglobulin receptor, impairs the epithelial immune barrier and allows microbes and inhaled particulates to activate innate immune signalling, resulting in persistent neutrophilic infiltration into the airway wall.28–30 In a mouse model of airway SIgA deficiency, we showed that neutrophil depletion blocks development of an age-related COPD phenotype, including reduced fibrotic remodelling of small airways and emphysema.31 Products of activated neutrophils, including reactive oxygen species and proteases such as neutrophil elastase and collagenase (matrix metalloproteinases 8), are likely contributors to ECM destruction in airway walls and lung parenchyma.32
While our studies implicated neutrophilic inflammation as a major factor in airflow obstruction and destruction of alveolar attachments in COPD, we also found that accumulation of other immune cells (CD4 T cells, CD8 T cells and B cell containing lymphoid follicles) correlate with reduced FEV1 in patients with COPD. In addition, increased numbers of CD8 T cells and B cell-containing follicles in and around small airway walls were associated with loss of alveolar attachments. The inverse correlation between B cell accumulation and alveolar attachments is consistent with a report by Tanabe et al 25; however, in this study the authors did not find increased numbers of T cells or neutrophils in terminal bronchioles from explanted lungs of patients with COPD compared with controls. Together, these findings raise the possibility that ongoing recruitment and activation of a variety of immune and inflammatory cells may contribute to airway pathology in COPD.
In addition to loss of alveolar attachments, other pathological lesions in small airways also appear to contribute to airflow limitation. Among these, altered airway epithelial cell differentiation towards a pseudostratified phenotype (also known as a goblet cell metaplasia) results in increased cellularity and epithelial height, which reduced the cross-sectional area of small airways by approximately 10% in patients with advanced COPD. In contrast, mucus plugging, another widely known finding in COPD, reduced luminal area by only ~3%.
Our study also provides information regarding the relationship of airway wall connective tissue remodelling and airflow limitation. While some studies have reported that subepithelial fibrosis and wall thickening are important pathological features of small airways in COPD,4 8 other studies have identified ECM degradation in airway walls of patients with COPD. Hogg et al reported increased collagen deposition in the subepithelium of small airways of patients with moderate COPD, but reduced collagen content with relative increase in collagen type III over collagen type I in small airways in patients with severe COPD.33 In addition, an increased proportion of immature/disorganised collagen type I fibres has been reported in COPD airways using a second harmonic generation approach.34 Our data support a two-step model in airway wall connective tissue remodelling during COPD progression. In FSs and patients with early COPD (GOLD stages I–II), increased collagen deposition occurs in airway walls, likely as the result of a chronic injury-repair process; however, ECM degradation predominates as the disease progresses to advanced stages (GOLD stages III–IV). This concept is further supported by our finding of widespread elastin fibre fragmentation, especially in outer zone of airway adventitia.
In COPD, major unmet therapeutic goals are interventions that would prevent or reverse the progressive airflow limitation. The present study highlights the importance of alveolar attachments as a tissue site at which such treatments may need to work. However, little is known about their development, maintenance programmes or capacity to repair following injury. These will be key questions for further study. A major challenge in the development of novel therapies for COPD has been to relate interventions to meaningful physiological effects. The current study, by providing evidence for a strong correlation between structure (alveolar attachments) and function (airflow), provides a strategic basis for future mechanistic and therapeutic studies.
Our study has several limitations, including that the archived tissue sections available were obtained from lungs that were not inflated prior to fixation. While normalising our morphometric measurements to RBM length allows comparable assessment of small airways, evaluation of surrounding lung parenchyma for emphysematous changes is difficult, thus limiting our ability to determine whether loss of alveolar attachments and emphysematous changes occur in close proximity. Also, because of the nature of these samples, we cannot address the possibility of regional variation within the lungs. Future studies to address regional relationships between emphysema (either by microCT or pathological evaluation) and loss of alveolar attachments would be beneficial, as well as studies quantifying alveolar attachments in patients with radiographic emphysema without a reduction in FEV1. Finally, we reported findings for each pathological feature of small airways as a composite measurement for each individual in this study and then summarised these data by disease category (NS, FS, COPD I–II, COPD III–IV); however, this reporting belies the heterogeneity of disease among patients with COPD. Rigorous pathological evaluation of these parameters in lungs from larger numbers of patients with COPD could shed additional light on the underlying heterogeneity of COPD.
We conclude that impaired integrity between small airways and adjacent alveolar tissue due to loss of alveolar attachments is a major factor responsible for declining lung function in COPD. Although small airway wall remodelling and luminal mucus plugging also contribute to airflow limitation, their relative contributions are less important. Ongoing activation of innate immunity with neutrophilic infiltration is a central feature of airway inflammation in COPD, which may promote airway wall pathology through damage to subepithelial connective tissue and loss of supporting alveolar attachments. New pharmacotherapies designed to inhibit these widespread destructive changes in small airways could limit disease progression in COPD.
Data availability statement
Research data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Lung specimens were originally collected with approval from the Institutional Review Board at Vanderbilt University Medical Center (protocol #s 060162 and 000616). This study was performed with deidentified specimens and was approved by the Institutional Review Board at Vanderbilt University Medical Center (protocol #161046).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors SSG, R-HD and VVP conducted the histopathological studies. PW and SSG performed statistical analysis. PPM provided human lung samples. KLS analysed CT scans. BWR, JMC and SR provided scientific and technical knowledge. VVP, SR and TSB cosupervised the project, overall design of the project and wrote the manuscript. All authors had final approval of the manuscript.
Funding This work was supported by grants from the US National Institutes of Health (NHLBI HL131906, NHLBI K08 HL138088, NCI CA152662), Department of Defense (W81XWH-17-1-0503) and Department of Veterans Affairs (l01BX002378, IK2BX00384).
Competing interests SR was an employee of AstraZeneca from 2015 to 2019 and continues to own shares that were received as part of his compensation. As part of that employment, he represented AstraZeneca on the Board of Directors of Dizal Pharma without additional compensation. In the last three years, he has consulted for Bergenbio, GlaxoSmithKline, NovoVentures and Verona. Between 1996 and 2007, his university received funding from tobacco companies that supported studies relating to harm reduction and to the impact of tobacco smoke on stem cells. As part of this work, he consulted with RJ Reynolds without personal fee on the topic of harm reduction, received funding from RJ Reynolds to evaluate the effect of a harm reduction product in normal smokers (1996) and in subjects with chronic bronchitis (1999) and to assess the effect of smoking cessation on lower respiratory tract inflammation (2000); he participated in a Philip Morris multicentre study to assess biomarkers of smoke exposure (2002); he received funding for a clinical trial from the Institute for Science and Health (2005), which receives support from the tobacco industry, to evaluate biomarkers in exhaled breath associated with smoking cessation and reduction. This study was supplemented with funding from Lorillard and RJ Reynolds to expand the spectrum of biomarkers assessed. He received a grant from the Philip Morris External Research Program (2005) to assess the impact of cigarette smoking on circulating stem cells in the mouse. There are no active tobacco-industry funded projects. All ties with tobacco industry companies and entities supported by tobacco companies were terminated in 2007.
Provenance and peer review Not commissioned; externally peer reviewed.