Article Text

Abstract
The lungs are exposed to a range of environmental toxins (including cigarette smoke, air pollution, asbestos) and pathogens (bacterial, viral and fungal), and most respiratory diseases are associated with local or systemic hypoxia. All of these adverse factors can trigger endoplasmic reticulum (ER) stress. The ER is a key intracellular site for synthesis of secretory and membrane proteins, regulating their folding, assembly into complexes, transport and degradation. Accumulation of misfolded proteins within the lumen results in ER stress, which activates the unfolded protein response (UPR). Effectors of the UPR temporarily reduce protein synthesis, while enhancing degradation of misfolded proteins and increasing the folding capacity of the ER. If successful, homeostasis is restored and protein synthesis resumes, but if ER stress persists, cell death pathways are activated. ER stress and the resulting UPR occur in a range of pulmonary insults and the outcome plays an important role in many respiratory diseases. The UPR is triggered in the airway of patients with several respiratory diseases and in corresponding experimental models. ER stress has been implicated in the initiation and progression of pulmonary fibrosis, and evidence is accumulating suggesting that ER stress occurs in obstructive lung diseases (particularly in asthma), in pulmonary infections (some viral infections and in the setting of the cystic fibrosis airway) and in lung cancer. While a number of small molecule inhibitors have been used to interrogate the role of the UPR in disease models, many of these tools have complex and off-target effects, hence additional evidence (eg, from genetic manipulation) may be required to support conclusions based on the impact of such pharmacological agents. Aberrant activation of the UPR may be linked to disease pathogenesis and progression, but at present, our understanding of the context-specific and disease-specific mechanisms linking these processes is incomplete. Despite this, the ability of the UPR to defend against ER stress and influence a range of respiratory diseases is becoming increasingly evident, and the UPR is therefore attracting attention as a prospective target for therapeutic intervention strategies.
- airway epithelium
- asthma
- viral infection
- innate immunity
- respiratory infection
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Endoplasmic reticulum stress and the unfolded protein response
The endoplasmic reticulum (ER) contains enzymes that modify newly synthesised proteins (eg, by disulphide bond formation) and chaperones to facilitate accurate three-dimensional folding. Excessive protein load, incorrect amino acid sequence or conditions that perturb protein folding lead to the accumulation of misfolded proteins. Sensors detect this ‘ER stress’ and initate signalling events that temporarily reduce global protein synthesis while enhancing production of folding chaperones. This unfolded protein response (UPR) also promotes the degradation of misfolded proteins (ER-associated protein degradation—ERAD), but if these compensatory mechanisms fail, the affected cells may undergo apoptosis.1 2
Misfolded proteins trigger the dissociation of the chaperone binding immunoglobulin protein ((BiP) also known as GRP78) from three transmembrane ER stress sensors: inositol-requiring kinase/endonuclease (IRE)1, protein kinase RNA-like ER kinase (PERK) and activating transcription factor (ATF)6, each of which initiates an arm of the UPR (figure 1).1 In addition, misfolded proteins may interact directly with IRE1 to trigger its activation, with BiP acting as a ‘sensitivity adjustor’ for different stressors.3 4 Protein synthesis is reduced by (a) IRE1-mediated cleavage of mRNAs (regulated IRE1-dependent mRNA decay—RIDD) and (b) PERK phosphorylation of the translation initiation factor eukaryotic translation initiation factor 2 alpha (eIF2α), which inhibits translation of most proteins.1

The unfolded protein response. A range of respiratory-relevant factors perturb protein homeostasis to activate endoplasmic reticulum (ER) stress sensors activating transcription factor (ATF) 6, inositol-requiring kinase/endonuclease (IRE) 1, and protein kinase RNA-like ER kinase (PERK). Activation of UPR signalling initiates downstream processes including protein and mRNA degradation via ER-associated protein degradation (ERAD) and regulated IRE1-dependent mRNA decay (RIDD), inhibition of global protein synthesis, upregulation of folding chaperones. If normal protein folding is not restored, apoptosis, inflammatory signalling and fibrotic responses may result, driving disease progression in a variety of conditions. CHOP, CCAAT homologous protein; eIF2α, eukaryotic translation initiation factor 2 alpha; PPP1, protein phosphatase 1; PM, particulate matter; ROS, reactive oxygen species; TLR, toll-like receptor; UPR, unfolded protein response, XBP, X-box binding protein.
Each ER stress sensor initiates the action of a transcription factor to induce UPR-dependent genes. IRE1 promotes splicing of X-box binding protein 1 (XBP1) mRNA to yield the active transcription factor XBP1s. ATF6 is cleaved to an active form in the Golgi. Active XBP1s and ATF6 localise to the nucleus and increase the transcription of UPR chaperone proteins such as BiP to improve folding capacity and components of the ERAD machinery to degrade misfolded proteins.1 Finally, phosphorylation of eIF2α by PERK paradoxically enhances the translation of some mRNAs, including the transcription factor ATF4 and its target CCAAT/enhancer-binding protein-homologous protein (CHOP).1 2 Together, ATF4 and CHOP induce protein phosphatase 1 regulatory subunit 15A (PPP1R15A), also known as GADD34, which selectively dephosphorylates eIF2α, providing a negative feedback loop.1 Because the recovery of translation mediated by PPP1R15A contributes to cell death during chronic ER stress, deletion of either CHOP or PPP1R15A renders cells more resistent to prolonged ER stress.5 The precise mechanisms by which ER stress initates apoptosis are incompletely understood; ER stress can trigger autophagy and alter mitochondrial bioenergetics to impact on cell fate in ways that appear to be context and cell-type specific.6–9 It is important to note that phosphorylation of eIF2α occurs in response to a range of cellular stresses including iron or amino acid deficiency.10 The downstream pathways involving ATF4 have therefore been termed the integrated stress response (ISR) and do not always equate to ER stress.10
ER stress triggers relevant to respiratory disease
Several inhaled triggers can activate local ER stress/UPR signalling. In some diseases UPR activation has been demonstrated in patient-derived lung tissue, with other studies undertaken in vitro or in animal models. Cell culture may not always fully recapitulate the multicellular airway environment, and species differences do exist. These factors may explain variation between studies and are flagged where relevant.
Cigarette smoke, particulate matter and asbestos
A range of inhaled agents provoke ER stress in airway epithelia and other relevant cells such as alveolar macrophages. Cigarette smoke (CS) contributes to the pathogenesis of COPD, lung cancer and possibly asthma and pulmonary fibrosis. Acute activation of the UPR by CS has been reported in airway epithelial cell lines and primary rodent and human bronchial epithelial cells.11–13 However, UPR-associated protein expression was not increased in mice subjected to chronic CS exposure or in airway epithelial cells from smokers vs non-smokers, but was increased in cells from patients with COPD, perhaps suggesting increased susceptibility to CS-induced ER stress in this patient group.14–17 CS may synergise with other environmental stressors, for example, prior CS-exposure increased UPR activation in the lungs of mice infected with respiratory syncytial virus (RSV).17
Mice exposed to fine particulate matter (PM2.5) displayed UPR activation in lung and liver, with similar effects seen in vitro in a PM2.5-treated macrophage cell line.18 Interestingly, the observed effects appeared to be mediated predominantly by the PERK-eIF2α axis. Although IRE1 was activated on PM2.5 exposure with increased RIDD, splicing of XBP1 mRNA was suppressed, an effect mirrored in human bronchial epithelial cells exposed to CS.13
Asbestos inhalation is linked to pulmonary fibrosis, mesothelioma and lung cancer. Treatment of the lung adenocarcinoma-derived A549 cell line and rat primary airway epithelial cells with asbestos increased expression of IRE1 and XBP1s mRNAs, and to a lesser extent proteins.19 However, direct detection of IRE1 and PERK protein is challenging, and the minor changes reported should be interpreted with caution. Of interest, increased expression of CHOP in human mesothelioma tissue was an independent predictor of poor survival, although the mechanism of this association is uncertain.20
These environmental agents simultaneously induce multiple cellular stress pathways, including ER stress and oxidative stress.18 19 21 ER stress and oxidative stress can interact, for example, reduced expression of the antioxidant enzyme glutathione peroxidase-1 in COPD airway cells was associated with an increased UPR and its restoration ameliorated UPR induction.17 Oxidation of redox-sensitive ER chaperones such as protein disulphide isomerase (PDI) represents a potential mechanism by which oxidative stress might promote ER stress.22
Bacterial, fungal and viral pathogens
UPR activation is induced by several pathogens relevant to respiratory disease including cystic fibrosis, asthma and COPD.
Toll-like receptor (TLR) activation and bacterial infection can trigger the UPR and may do so selectively. For example, TLR2 and TLR4 specifically activated IRE1 to promote inflammatory mediator release.23 24 Respiratory pathogens may also subvert the UPR; Legionella pneumophila, an intracellular pathogen that replicates in an ER-associated compartment, selectively blocks activation of the IRE1 pathway.25 Secreted bacterial toxins can modulate the UPR; for example, pyocyanin (from Pseudomonas aeruginosa) triggered this response in human primary airway cells, evidenced by XBP1 splicing and induction of BiP.26 It is worth noting that induction of PPP1R15A, a protective response in this study, was more dependent on the ISR than PERK, highlighting the importance of fully dissecting stress-sensitive pathways and not confusing ISR activation with the UPR.
Aspergillus fumigatus is a fungal pathogen that interacts variably with airway defences, leading to a range of outcomes including allergic bronchopulmonary aspergillosis (ABPA). BiP expression was increased in lung tissue from patients with ABPA.27 Administration of A. fumigatus to mice induced pulmonary UPR and airway hyperresponsiveness. The mechanism(s) remain to be fully elucidated but involved generation of mitochondrial reactive oxygen species (ROS) and disruption of PDI function, reminiscent of the findings in CS-induced ER stress.22 27 28
Many viruses induce ER stress including RSV, influenza A virus (IAV), coxsackievirus A16, SARS-CoV-1 and SARS-CoV-2 (the causes of SARS and COVID-19 respectively).29–33 The mechanism of viral-induced ER stress may relate to prolific viral protein translation overwhelming protein folding capacity.26 34 IAV was reported to activate IRE1 with little activation of PERK or ATF6, leading to inflammation and apoptosis of primary human bronchial epithelial cells.31 It is worth noting, however, that UPR sensors show differing time courses of activation and inactivation. In this study, PPP1R15A was induced, raising the possibility that transient PERK activation might have been missed. Treatment with tauroursodeoxycholic acid (TUDCA), a compound with complex effects including the amelioration of ER stress or with 3,5-dibromosalicylaldehyde (a putative IRE1 inhibitor) significantly reduced viral replication. While such observations raise the possibility that enhanced ER stress promotes IAV replication, they should be strengthened by appropriate genetic knockout experiments. Picornaviruses, such as rhinovirus, might also benefit from activation of IRE1, since this may promote autophagy and picornaviruses use autophagosomes as sites of RNA replication.35 In contrast, while RSV was also reported to induce non-canonical activation of the UPR with activation of IRE1 and ATF6 but not PERK, IRE1 suppressed RSV replication, suggesting that inhibiting this arm of UPR could be detrimental in RSV infection.36
Thus infection may activate the UPR pathways in a pathogen-specific fashion, and host responses may counter pathogen survival/replication or be suborned to promote it.
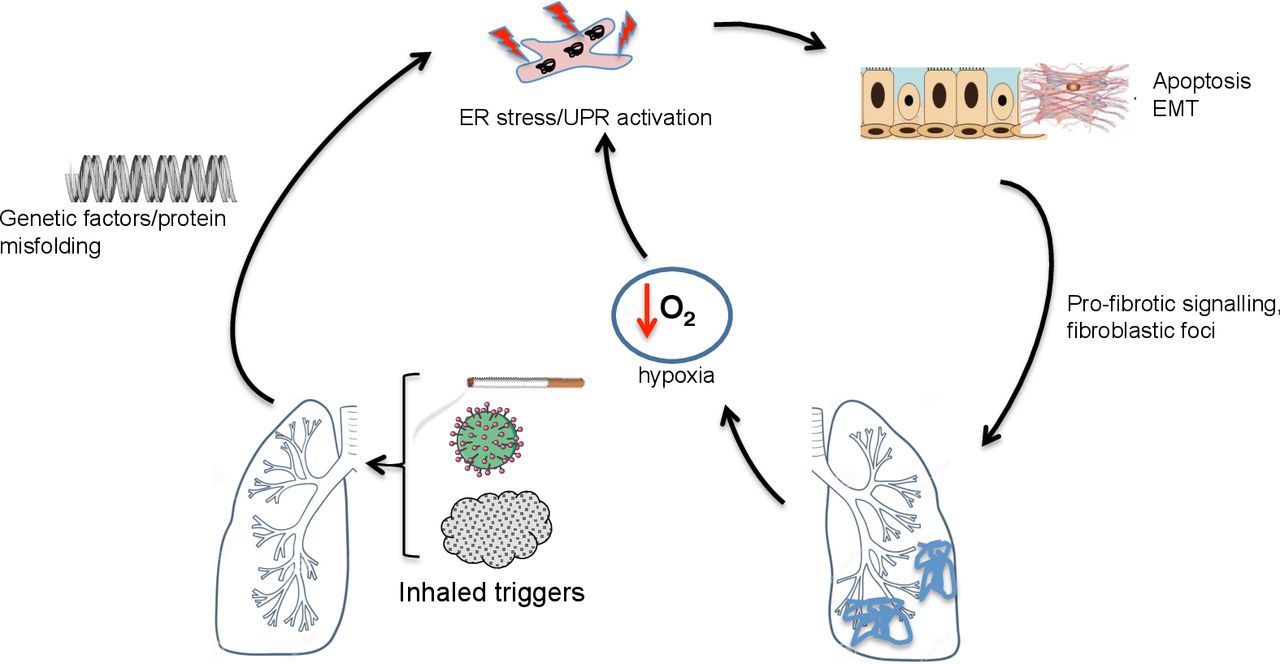
Hypoxia
Systemic hypoxia results from any severe lung disease, and profound tissue hypoxia characterises inflamed or poorly vascularised tissues; examples include COPD and CF airways, or the tumour microenvironment. Links between hypoxia and UPR have been extensively investigated, particularly in the context of cancer.37 While hypoxia triggers the UPR in a range of targets including airway epithelial cells, the mechanism(s) by which this occurs is/are incompletely understood.38 ROS generated in the context of hypoxia may modulate UPR activation by direct or indirect effects on BiP and by impaired disulphide bond formation.39 40 The redox-sensitive chaperone PDI becomes reduced during protein folding; it recovers by transferring electrons to ERO1 (ER oxidoreductin), which then requires molecular oxygen to become reoxidised and regain function.41 Furthermore, ERO1 is a hypoxia-inducible factor (HIF) target, hence hypoxia might regulate disulphide bond formation by multiple routes.42 Several pathways thus exist by which hypoxia may induce ER stress and UPR, and compensatory pathways attempt to mitigate their impact and restore cellular homeostasis.
These factors together with host-specific predispositions may provoke ER stress/UPR in a disease-specific context. Exploitation of these pathways is increasingly viewed as a therapeutic possibility in treatment-recalcitrant conditions. A fuller understanding of the role of UPR in respiratory diseases is thus an important research topic.
The UPR and pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is a scarring disease, characterised histologically by fibroblastic foci. Despite antifibrotic therapies, prognosis remains poor.43 The biological mechanisms that drive this condition remain unclear; the study of rare familial cases has suggested ER stress plays an important role (figure 2).

ER stress and the unfolded protein response in pulmonary fibrotic lung disease. In pulmonary fibrosis, triggers such as cigarette smoke, asbestos, particulate matter or viruses may trigger ER stress in a genetically predisposed (eg, surfactant protein mutations, MUC5B polymorphisms) host. This is associated with apoptosis and epithelial mesenchymal transition (EMT). Tissue hypoxia may further promote ER stress in affected areas. ER, endoplasmic reticulum; MUC5B, mucin 5B; UPR, unfolded protein response.
Alveolar type II (AT2) cells secrete surfactant; mutations in surfactant protein C (SFTPC) have been associated with familial IPF and profound UPR activation.44 Many of these mutations disrupt the SFTPC BRICHOS domain, which acts as a self-chaperone to facilitate SFTPC folding.45 Impaired SFTPC folding can lead to protein aggregation and activation of all arms of the UPR.46 In mouse models, these mutations either disrupt lung morphogenesis to drive fibrosis directly or sensitise the lung to ‘second hits’ that induce pulmonary fibrosis.47 48
Host factors have also been associated with IPF in genome-wide association studies (GWAS), and UPR provides a potential link between these associations and disease. The MUC5B promoter variant rs35705950 (associated with increased MUC5B (mucin 5B) expression) is the strongest population-wide genetic risk factor associated with IPF.49 XBP1s is coexpressed with MUC5B in IPF lungs, induces expression of MUC5B in the distal bronchiolar epithelia and preferentially activates the rs35705950 promoter variant, providing a potential positive feedback mechanism linking UPR-induced expression of MUC5B and IPF.50 Precisely how MUC5B might promote IPF remains uncertain and warrants further investigation.
ER stress is also implicated in sporadic IPF. Deletion of BiP from murine AT2 cells predictably not only resulted in activation of the UPR but also led to spontaneous age-related pulmonary fibrosis.51 Markers of UPR are present in fibrotic lungs from patients with sporadic as well as genetically determined IPF.52–54 Upregulation and activation of ATF6, prominent expression of ATF4 and CHOP and activation of the IRE1 pathway were observed in alveolar epithelial cells in fibroblastic foci; these markers colocalised with cleaved caspase 3 and TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) staining, suggesting UPR is linked to apoptosis in this setting.52
Epithelial-mesenchymal transition (EMT) has also been implicated in IPF pathogenesis with several studies describing ER stress-induced EMT; upregulation of UPR markers XBP1s and BiP and myofibroblast marker α-smooth muscle actin, coincided with a reduction in epithelial cell markers including zonula occludens-1 and E-cadherin and a fibroblast-like morphology.55 56
Additional links between UPR and pulmonary fibrosis exist. As noted, CS and particulates activate the UPR; smoking is a risk factor for IPF and ambient particulates are associated with accelerated decline in this disease.57 Hypoxia-related signalling may exacerbate ER stress locally: HIF-1α and CHOP proteins were detected together in alveolar epithelium in patients with IPF lung biopsies, in murine bleomycin-induced pulmonary fibrosis and in epithelial cells from rats exposed to acute hypoxia to initiate fibrosis.58 As noted above, viral infections can induce ER stress, and viral infections may initiate IPF exacerbations. Both herpes viral DNA and antigens have been associated with IPF lung tissue, the latter colocalised with markers of activated UPR.59
Although most studies have focused on IPF, UPR activation has been reported in other pulmonary fibrotic conditions. For example, markers of UPR activation were found in the proteome of lungs from patients with fibrotic nonspecific interstitial pneumonia (NSIP).52 UPR was also detected in macrophages exposed to asbestos in vitro, and in the alveolar macrophages of mice exposed to asbestos and humans with asbestosis.60 In a mouse model of amiodarone-induced pulmonary fibrosis, surfactant accumulation was observed together with induction of ER-stress and epithelial apoptosis.61 Although these reports are limited in number, they suggest that ER-stress and UPR may be a common pathway in the induction and/or progression of fibrotic lung disease including but not limited to IPF.
The UPR and asthma
GWAS have linked ORMDL3 with asthma. ORMDL3 regulates both ceramide biosynthesis and UPR activation; these pathways could interact to promote changes in airway physiology during allergic responses.62 In mice, ORMDL3 is an allergen-inducible airway epithelial protein, and ORMDL3 knockout mice were protected from Alternaria-induced allergic airway disease, with decreased activation of ATF6/XBP1.63
Overexpression of ORMDL3 selectively activated ATF6 in A549 cells and increased expression of phosphorylated eIF2α and BiP in HEK293 cells, contrasting with depletion of these markers by treatment with ORMDL3 siRNA.64 65 However, in a separate study, knockdown of ORMDL3 did not affect phosphorylated eIF2α, CHOP or spliced XBP1 expression in A549 or 1HAE airway epithelial cells.66 Effects may thus be cell-type or species-specific or relate to over-expression and remain to be investigated in primary asthmatic airway cells.
Murine models have suggested a role for UPR in asthma pathogenesis, with upregulation of UPR markers (BiP, CHOP, ATF6, XBP1s and phosphorylated eIF2α) in lung tissue from ovalbumin (OVA)-sensitised or OVA-LPS (lipopolysaccharide)-sensitised mice.67 68 Induction of BiP and CHOP has been shown in blood monocytes and alveolar macrophages from human asthmatics.67 ER stress has also been associated with the expression of interferon-stimulated genes and type 2 inflammation in airway epithelial brushings from mild asthma.69 Asthma exacerbations are often driven by respiratory viruses known to induce ER stress, which could lead to repeated cycles of UPR activation, and thus contribute to sustained inflammation and airway remodelling. In OVA-induced ‘asthma’ in mice, pretreatment with the compound 4-phenylbutryic acid (4-PBA, which has a range of actions that include alleviation of ER stress, described below) led to a significant reduction of UPR markers BiP and CHOP, with concomitant reduction in airway inflammation and bronchial hyper-responsiveness.67 68 Importantly, established asthmatic features were also attenuated by 4-PBA administered after completion of OVA challenge.67 House dust mite (HDM) models have also demonstrated induction of UPR,70 and ER stress markers were upregulated in mice in vivo and human bronchial and nasal epithelial cells challenged with HDM in vitro.71 72 As noted above, inhaled A. fumigatus has been linked to UPR activation and ABPA in mouse and human studies.22 27 28
Overall, these studies imply a link between ER stress in asthma and suggest that aberrant UPR activation may contribute to asthma pathogenesis or progression in some settings (see figure 3).

ER stress and the unfolded protein response in asthma. In asthma, triggers such as house dust mite, viral or fungal pathogens, and cigarette smoke may trigger ER stress in a genetically predisposed (eg, ORMDL3 polymorphisms) host. This is associated with inflammation, apoptosis, mucus hypersecretion and leads to airway reactivity and remodelling. ER, endoplasmic reticulum; UPR, unfolded protein response.
The UPR and COPD
Despite extensive GWAS and sequencing studies in COPD, no clear genetic links have emerged with ER stress or UPR. CS can induce ER stress in airway epithelial cells, but this does not equate to a mechanistic link, and multiple cell stress pathways are initiated by CS. Min and colleagues reported increased expression of phosphorylated eIF2α and CHOP by immunoblotting in the lungs of patients with COPD, although the IRE1 and ATF6 arms of the UPR were not investigated.73 However, a separate study detected no increase in ER stress markers (including ATF4, ATF6 and CHOP) quantified by western blotting and PCR in the lungs of patients with COPD.54 These differences might reflect differing methodologies or patient heterogeneity, but additional studies are needed to clarify such discrepancies. Hassan and colleagues reported that decreased expression of miR199a-5p underpinned increased expression of BiP, ATF6 and XBP1s in patient with COPD monocytes, an intriguing finding but with uncertain significance to lung pathology.74
Mouse models have also suggested possible roles for ER stress in emphysema, although how such models recapitulate human disease is debatable. 4-PBA was partially protective in a mouse model of CS-induced emphysema, and mice treated with the heme-scavenging protein haemopexin developed less ER stress, airway fibrosis and less emphysema than control mice in response to inhalation of bromine.15 75 Studying the development of COPD in mice with targeted mutations in UPR genes in such models should yield more definitive results.
Thus, overall, while UPR may be activated in the context of COPD, the contribution of UPR to COPD pathogenesis is currently unclear.
The UPR and cystic fibrosis
Loss-of-function mutations affecting the cystic fibrosis transmembrane conductance regulator (CFTR) cause CF. Class II CFTR mutations including F508del (present in approximately 90% of patients with CF) lead to protein misfolding. Misfolded F508del-CFTR fails ER quality control and is targeted for proteasomal degradation, with reduced cell membrane delivery.76 However, most CFTR mutations (including F508del) reside in cytoplasmic domains, and hence would not be expected to induce the UPR. Studies of the role of UPR in CF airway epithelia are complicated by the use of overexpressed versus endogenous protein, primary cells versus cell lines and the presence of airway infection.
Overexpression of F508del activates UPR, with increased expression of ATF6 and BiP; however, protein overexpression per se may induce ER stress and this finding is non-specific.77 78 Atypical activation of UPR with enhanced XBP1 splicing was reported in CF lung tissue and macrophages versus non-CF samples, but the airway cell work supporting this observation lacked appropriate control cells.79 Ribeiro and colleagues also demonstrated increased levels of XBP1s in native CF compared with normal human airway epithelia, with enhanced secretion of inflammatory mediators; importantly, they found that infected CF secretions induced equivalent XBP1 splicing and increased BiP expression, demonstrating that induction of UPR in CF epithelia is likely secondary to infection rather than CFTR misfolding.80 81 Similar findings were reported in alveolar macrophages from patients with CF versus healthy controls, with enhanced XBP1 splicing suggested to be secondary to the inflammatory milieu rather than to CFTR misfolding.82 Enhanced activation of the IRE1α-XBP1 pathway in CF monocytes and macrophages was associated with increased glycolysis and mitochondrial function, plus exaggerated TNF (tumour necrosis factor) and IL-6 production, suggesting relevant functional consequences.83
Thus, ER stress may occur in the context of CF and have proinflammatory actions, however, it likely reflects airway infection rather than a primary effect of protein misfolding (see figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
ER stress and the unfolded protein response in cystic fibrosis. In cystic fibrosis, pulmonary infection leads to ER stress and UPR activation in airway epithelial cells and macrophages, promoting inflammatory injury and hence further increasing the potential for infection. ER, endoplasmic reticulum; UPR, unfolded protein response. TNF, tumour necrosis factor; IL6, interleukin 6.
The UPR and lung cancer
The role of ER stress and the UPR is well established in a range of cancers. Solid tumours are oxygen‐deprived and nutrient‐deprived environments, factors that will promote ER stress. Increased expression of BiP, phosphorylated eIF2α and XBP1s has been demonstrated in several lung cancer studies.13 84 85 Ribosome-binding protein 1 (RRBP1) is highly expressed in lung cancer tissue and may help render cancer cells tolerant of ER-stressful environments by enhancing BiP expression.84–86Accordingly, knockdown of RRBP1 lowered BiP levels and reduced the tumorogenic potential of lung cancer xenografts, regulating apoptosis via activation of the p38/JNK pathway .87
Several studies implicating UPR in lung cancer treatment-resistance have been performed in the A549 lung adenocarcinoma cell line; additional evidence is required to confirm that these effects are clinically relevant. Some researchers have employed models with greater disease relevance. Using patient-derived xenografts, KRAS-mutant lung cancer cells resistant to pemetrexed and trametinib were found to be highly dependent on UPR for survival, with a selective vulnerability to drug combinations including targeting the UPR.88 A recent paper implicated a novel post-translational pathway in linking ER stress to treatment resistance: deletion of the ufmoylation pathway triggered a protective UPR in lung cancer ‘persister cells’ tolerant to tyrosine kinase inhibitors, associated with protumorigenic inflammatory signalling. Thus, post-translational modifications may regulate UPR signalling independent of protein expression and may warrant further investigation in lung cancer and other conditions.89 90
The role of the UPR in lung cancer is currently unclear but understanding is increasing and may for example enable ‘synthetic lethality’ approaches to target drug-resistant targets.
UPR and potential theraputic intervention in respiratory disease
While blocking the UPR should be approached with caution due to the homeostatic nature of this system, uncovering the complexity and individuality of UPR activation in different disease states has generated interest in targeting specific UPR components, developing ‘chemical chaperones’ and utilising modulators of ERAD (see table 1).1
Therapeutic intervention of UPR and ER stress
Evidence that airway diseases such as asthma and emphysema respond to pharmacological manipulation of the UPR is limited to interventions in animal and cellular models. For example, salubrinal (which promotes phosphorylation of eIF2α by poorly understood mechanisms) protected cultured airway epithelial cells from CS-induced apoptosis and repressed CS‐induced airway epithelial ER stress in a mouse model, with the mice protected from the development of emphysema.73 75 91 92 Mammalian target of rapamycin (mTOR) inhibitor rapamycin prevented ER stress-induced apoptosis through activation of autophagy and removal of misfolded proteins in vitro.93 Mediators downstream of mTOR have been used to modulate UPR. For example, AMP-activated protein kinase (AMPK) activators, including metformin, reduced palmitate-induced and LDL-induced ER stress in a diabetes disease context.94 Peroxisome proliferator-activated receptor agonists have also been shown to inhibit palmitate-induced UPR in vivo and in vitro.94 In cancer, a range of UPR modulating factors that target ER stress proteins has been described, including PERK and IRE1 inhibitors.94 Chemical chaperones such as 4‐PBA and TUDCA have been used widely in animal models, for example, ameliorating ER stress, inflammation and fibrosis in bleomycin-induced pulmonary fibrosis and attenuating ER stress and manifestations of allergen-induced ‘asthma’ in mouse models.67 68 95–97 A note of caution should be sounded, as the term ‘chemical chaperones’ is often used for small molecules such as 4-PBA and TUDCA, but there is little evidence they function through direct interaction with misfolded proteins. Rather, they appear to affect a complex repertoire of cellular processes, whose relevance to ER stress is uncertain. For example, 4-PBA has also been shown to modulate cargo sorting into COPII vesicles and to reduce nuclear translocation of NF-κB.67 98 TUDCA and another conjugated bile acid, alanyl β-muricholic acid, have recently been suggested to bind ATF6 to inhibit HDM-induced UPR and allergic airway disease, but bile acids have other functions, notably altering gene transcription via receptors such as the nuclear farnesoid X receptor.99 Inhibitors of ER stress sensors such as PERK are being developed and may also have off-target effects.100 Ongoing initiatives to develop more specific compounds should both enhance our understanding of the consequences of ER stress and UPR and offer more targeted therapeutic opportunities.
Conclusions
Many respiratory insults are associated with ER stress, but causality cannot be assumed. There is reasonable evidence for a causal role of ER stress and the UPR in a restricted range of lung diseases, such as familial and perhaps sporadic IPF, infection in CF and treatment-resistance in certain lung cancers. However, further work is required to improve our understanding of its role in conditions such as asthma, while evidence for a role in COPD is currently limited to correlative studies. Genetic models may yield more definitive mechanistic insights than use of poorly characterised small molecule inhibitors. A better understanding of how such compounds restrain cellular stress responses may facilitate the development of treatments targeting UPR alone, or individual UPR components that are dysfunctional or hijacked in specific respiratory disease states. The therapeutic potential of the UPR is thus an exciting prospect for the future, but more work is required before this promise can be fulfilled.
References
Footnotes
Twitter @MarciniakLab
SJM, LCP and AMC contributed equally.
Contributors The article was conceived by KLB, CAS, SJM, LP and AMC. KLB and AMC were responsible for the initial draft preparation and revision. All authors were involved in reviewing each draft and approving the final manuscript. AMC, SJM and LP are responsible for the overall content as guarantors.
Funding KLB is funded by the Rosetrees Trust.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.