Article Text

Abstract
Introduction Idiopathic pulmonary fibrosis (IPF) is a progressive fibrosing interstitial lung disease of unknown aetiology and cure. Recent studies have reported a dysregulation of exosomal microRNAs (miRs) in the IPF context. However, the impact of IPF-related exosomal miRs on the progression of pulmonary fibrosis is unknown.
Methods Two independent cohorts were enrolled at the ambulatory care polyclinic of Liège University. Exosomes from sputum were obtained from 19 patients with IPF and 23 healthy subjects (HSs) (cohort 1), and the ones from plasma derived from 14 patients with IPF and 14 HSs (cohort 2). Exosomal miR expression was performed by quantitative reverse transcription–PCR. The functional role of exosomal miRs was assessed in vitro by transfecting miR mimics in human alveolar epithelial cells and lung fibroblasts.
Results Exosomal miR analysis showed that miR-142-3p was significantly upregulated in sputum and plasma of patients with IPF (8.06-fold, p<0.0001; 1.64 fold, p=0.008, respectively). Correlation analysis revealed a positive association between exosomal miR-142-3p and the percentage of macrophages from sputum of patients with IPF (r=0.576, p=0.012), suggesting macrophage origin of exosomal miR-142-3p upregulation. The overexpression of miR-142-3p in alveolar epithelial cells and lung fibroblasts was able to reduce the expression of transforming growth factor β receptor 1 (TGFβ-R1) and profibrotic genes. Furthermore, exosomes isolated from macrophages present antifibrotic properties due in part to the repression of TGFβ-R1 by miR-142-3p transfer in target cells.
Discussion Our results suggest that macrophage-derived exosomes may fight against pulmonary fibrosis progression via the delivery of antifibrotic miR-142–3 p to alveolar epithelial cells and lung fibroblasts.
- Idiopathic pulmonary fibrosis
- Interstitial Fibrosis
- Airway Epithelium
- Macrophage Biology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is the key question?
What is the impact of idiopathic pulmonary fibrosis (IPF)-related exosomal microRNA (miR)-142-3p on the progression of pulmonary fibrosis?
What is the bottom line?
Correlation analysis revealed that the expression of sputum exosomal miR-142-3p is positively associated with sputum macrophages of patients with IPF, suggesting its macrophage origin.
Macrophage-derived exosomes present antifibrotic properties due in part to the transfer of miR-142-3p to alveolar epithelial cells and lung fibroblasts.
Why read on?
This study provides novel insights into the potential protective role of macrophage-derived exosomes in IPF context.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrosing interstitial lung disease of unknown aetiology and cure, which leads to rapid death, within 2–3 years after diagnosis.1–4 IPF is characterised by progressive and irreversible destruction of the lung architecture caused by fibrotic ‘scar’ formation that ultimately leads to organ destruction and death from respiratory failure.5 6 Its physiopathology remains poorly characterised, although recent studies suggest that this disease results from aberrant dysregulated wound healing response following chronic alveolar epithelial injury and aberrant proliferation of fibroblasts.7 Genetic predispositions and environmental factors (such as smoking, gastro-oesophageal reflux), as well as cell senescence associated with ageing appear to be key factors in IPF pathophysiology.8–10 Its clinical prognosis has recently improved since the discovery of new efficient therapeutic agents, pirfenidone and nintedanib.2 11 Yet, these therapies only slow disease progression and do not provide a definitive cure. Novel therapeutics are desperately needed.
MicroRNAs (miRs) are small non-coding RNA molecules (20–22 nucleotides) that post-transcriptionally modulate gene expression by regulating the stability and/or translation of target messenger RNAs (mRNAs) by binding to the 3′ untranslated region of mRNAs.12 13 MiRs have emerged as potent modulators of various cellular processes such as proliferation, migration, differentiation, inflammation and their utility as biomarkers of pulmonary fibrosis is being explored.14–18 Indeed, Lui et al found an upregulation of miR-21-5p in the lungs of patients with IPF and its profibrotic properties by promoting the activity of transforming growth factor β1 (TGF-β1) in pulmonary fibroblasts.19 On the other hand, other studies reported that the levels of miR-200c-5p,20 miR-26a-5p,21 miR-29-5p22 and Let-7d-5p23 were reduced in the lungs of mice with experimental pulmonary fibrosis and/or in the lungs of patients with IPF, and their overexpression attenuates experimental pulmonary fibrosis in mice. However, the role of miRs in the pathogenesis of lung fibrosis has just begun to be elucidated. Further studies on the role of these molecules on lung fibrosis could help us to develop new therapeutic strategies for IPF.
Exosomes are small vesicles (30–150 nm) that are constitutively secreted into biofluids (eg, blood24 and bronchoalveolar lavage fluid25) by several cell types, including alveolar epithelial cells, fibroblasts and inflammatory cells, and contain numerous bioactive molecules, such as nucleic acids (including miRs), proteins and lipids.26 Their content varies according to the state of their parental cells and is thus reflective of the cellular context.27 Several studies reported that exosomes regulate various signalling pathways, including inflammatory and angiogenic pathways, by transferring miRs from a donor to a recipient cell.28–30 Exosomes and miRs appear to contribute to pulmonary fibrosis, as the size, quantity, content and function of exosomes vary with inflammation and epithelial damage,27 31 and several miRs are known to modulate fibrosis pathways.19–22 A limited number of studies have reported the role of exosomes on the pathogenesis of pulmonary fibrosis.17 31 32 However, to our knowledge, no study has reported the impact of exosomal miRs derived from sputum on the pathogenesis of pulmonary fibrosis.
Previously, we have identified three miRs showing an aberrant expression in sputum-derived exosomes from patients with IPF compared with healthy subjects (HSs) (miR-142-3p, miR-33a-5p and let-7d-5p).33 In the current study, we sought to determine the impact of these miRs on fibrosis associated to IPF progression. Our results suggest that (1) sputum macrophages are the source of elevated levels of exosomal miR-142-3p in IPF context; and (2) macrophage-derived exosomes exert a protective effect against pulmonary fibrosis progression via the delivery of antifibrotic miR-142-3p.
Methods
Demographic and clinical characteristics of cohorts
Two independent study populations were enrolled at the ambulatory care polyclinics of Liège University. The diagnosis of (definite) IPF was made according to the international recommendations of the American Thoracic Society/European Respiratory Society34 (more information in online supplementary methods). The first cohort of 19 patients with IPF and 23 HSs was used to study the alteration of exosomal miR levels in the sputum, whereas the second cohort of 14 patients with IPF and 14 HSs was used to study the alteration of exosomal miR levels in the plasma. Tables 1 and 2 provide an overview of the demographic and clinical characteristics of the 42 subjects from the first cohort (sputum cohort) and 28 subjects from the second cohort (plasma cohort). In both cohorts, patients with IPF exhibit a restrictive respiratory pattern.
Supplemental material
Demographic and clinical characteristics of patients with HS and IPF enrolled in the sputum-exosomal microRNA study (cohort 1)
Demographic and clinical characteristics of patients with HS and IPF enrolled in plasma-exosomal microRNA study (cohort 2)
Sputum induction and processing
After premedication with 400 μg inhaled salbutamol, sputum was induced by inhalation of hypertonic (NaCl 5%) or isotonic (NaCl 0.9%) saline according to the forcedexpiratory volume in one second value (greater than or less than 65% predicted). The whole sputum was collected in a plastic container, weighed and homogenised by adding three volumes of phosphate-buffered saline, vortexed for 30 s and centrifuged at 800 g for 10 min at 4°C. The supernatant was separated from the cell pellet and stored at −80°C.35
Plasma processing
Blood was collected and transferred to EDTA-containing tubes. Plasma was isolated from the blood by centrifugation at 1500 g for 10 min at 4°C to remove blood cells, then the supernatant was centrifuged at 3000 g for 15 min at 4°C to remove platelets and cell debris. Plasma was stored at −80°C.
Isolation of exosomes from biofluids (sputum and plasma) or culture medium, dynamic light scattering, western blotting, quantitative reverse transcription–PCR (qRT-PCR), cell culture/treatments/transfection/5-Bromo-2'-Deoxyuridine (BrdU) assay and in silico analysis.
Detailed methodology can be found in the online supplementary methods.
Statistical analysis
Statistical analysis was performed using SPSS Statistics for Windows software V.20 or GraphPad Prism V.8. A p value of less than 0.05 was considered to be statistically significant. For the analysis of subject characteristics, the values are expressed as mean±SD or median (IQR). Comparisons between groups were performed either with parametric unpaired two-tailed Student’s t-test or non-parametric two-tailed Mann-Whitney test. Differences in categorical data were compared using χ2 test. For comparison of miR expression in exosomes from patients with IPF versus HSs, data were not-normally distributed and are analysed using non-parametric two-tailed Mann-Whitney test, followed by Bonferroni-Dunn correction. P values were adjusted for age and sex using the general linear model; age and sex were entered into the analysis as covariates. For correlation analysis between miR levels and sputum cell counts (%), data were not normally distributed and analysed unsing Spearman’s correlation. Individual data points are shown in all plots and represent data from independent cell culture experiments. For cell culture experiments, normality distribution was assessed with Shapiro-Wilk normality test. Variables with normal distribution are summarised as mean (±SEM) and with statistical comparison between two groups performed using paired two-tailed Student’s t-test.
Results
Exosomal miR-142-3p levels are increased in induced sputum and plasma of patients with IPF
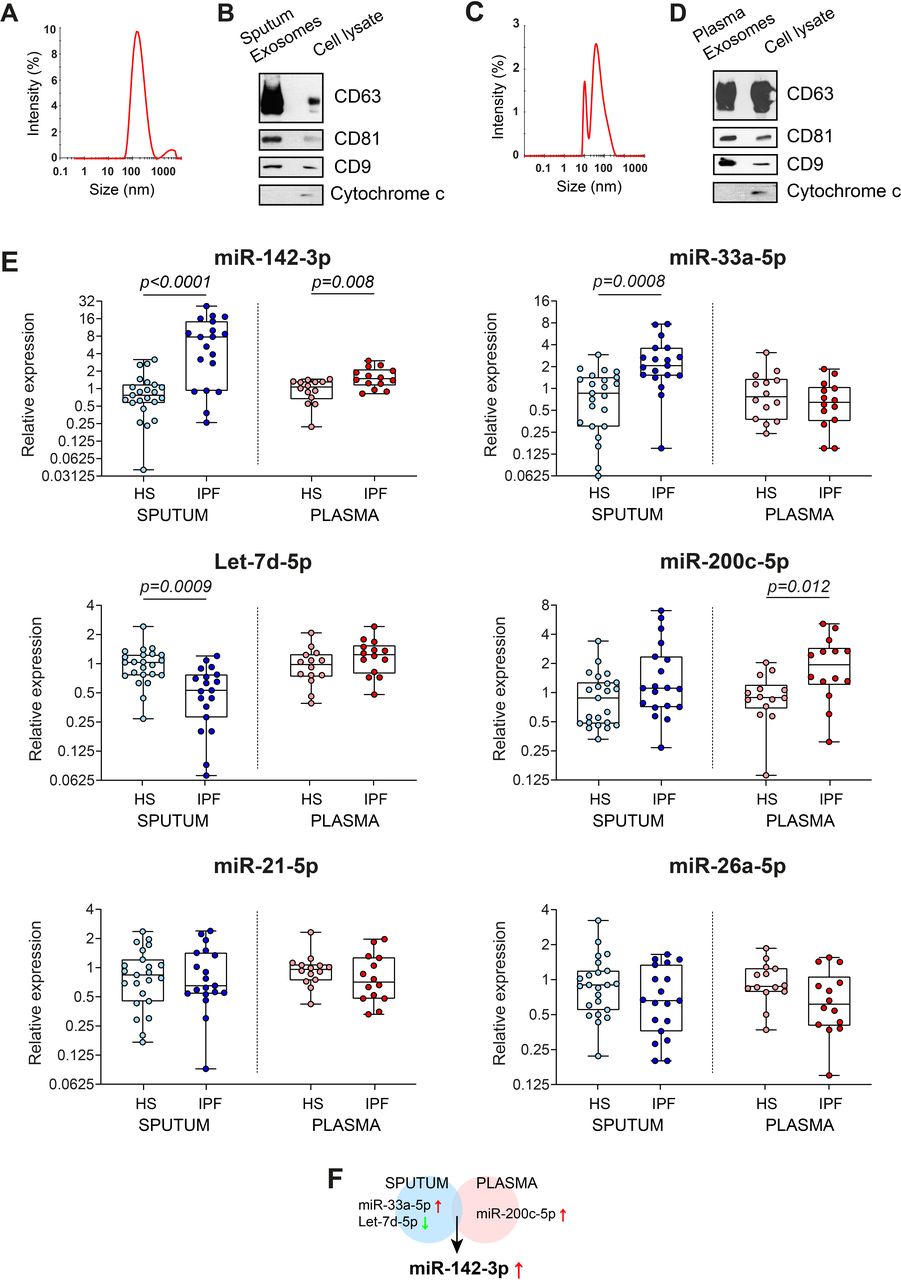
Previously, we identified a unique signature of three miRs (miR-142-3p, miR-33a-5p and Let-7d-5p) as potential biomarkers for IPF from sputum-derived exosomes.33 Here, we sought to determine whether these miRs are also dysregulated in the plasma of patients with IPF. For this, we first increased the sample size of the sputum cohort by isolating exosomes from the sputum of nine HSs and three patients with IPF using standard ultracentrifugation protocol (as described in Njock et al 33) for a total of 23 HSs and 19 patients with IPF. In parallel, we isolated exosomes from the plasma of 14 HSs and 14 patients with IPF. The demographic and clinical characteristics of the subjects were listed in table 1 (sputum cohort) and table 2 (plasma cohort). The isolated vesicles from sputum and plasma had an average size distribution of 140 nm (±19.1) (figure 1A) and 51.8 nm (±10.2) (figure 1C), respectively, typical size distribution of exosomes. Furthermore, these isolated vesicles present an enrichment of several exosomal markers, CD63, CD81 and CD9, and an absence of mitochondrial cytochrome c (figure 1B,D), which confirm the purity of our sputum-derived and plasma-derived exosome preparations.

miR-142-3p is upregulated in sputum-derived and plasma-derived exosomes of patients with IPF compared with HS. (A,C) Representative size distribution of purified exosomes from (A) sputum and (C) plasma by dynamic light scattering analysis. A representative experiment of 3 is shown. (B,D) Western blot analysis of exosomal markers, CD63, CD81, CD9, and mitochondrial protein cytochrome c in lysates from (B) sputum-derived and (D) plasma-derived exosomes and cell lysate (A549 cells). (E) Quantitative real-time PCR analysis of specific miRs in sputum-derived and plasma-derived exosomes of patients with IPF (n=19 for IPF sputum, n=14 for IPF plasma) compared with HS (n=23 for HS sputum, n=14 for HS plasma). Data are non-normally distributed and are analysed using a non-parametric two-tailed Mann-Whitney test, followed by Bonferroni-Dunn correction. (F) Venn diagram of the dysregulated miRs in sputum and plasma of patients with IPF. miRs in blue and red circles represent the sputum-deregulated and plasma-deregulated miRs, respectively. HS, healthy subject; miR, microRNA; IPF, idiopathic pulmonary fibrosis.
Then, we compared the expression levels of six IPF-related miRs in induced sputum and plasma between patients with IPF and HSs. For this, we chose to study six IPF-related miRs identified previously in sputum (miR-142-3p, miR-33a-5p and Let-7d-5p)33 and plasma/serum (miR-21-5p,14 16 miR-200c-5p20 or miR-26a-5p21) of patients with IPF. We confirmed an alteration of miR-33a-5p (2.73 fold, p=0.0008) and Let-7d-5p (−1.78 fold, p=0.0009) in IPF-sputum exosomes, and miR-200c-5p (2.21 fold, p=0.012) in IPF-plasma exosomes compared with corresponding HS-exosomes (figure 1E). Interestingly, only miR-142-3p was dysregulated similarly in sputum-derived and plasma-derived exosomes from patients with IPF compared with HS (figure 1E). The elevation of exosomal miR-142-3p is more pronounced in the sputum (8.06-fold, p<0.0001) than the plasma (1.64 fold, p=0.008) of IPF patients (figure 1E). The results were also significant after adjusting for age and gender distribution (table 3).
General linear model to adjust p values for age and gender of exosomal miR expression
Our results show that among the six miRs that we investigated, only miR-142-3p is altered in exosomes from sputum and plasma of IPF patients (figure 1F), suggesting its major role in IPF pathophysiology.
Cellular origin responsible for the elevation of exosomal miR-142-3p in IPF sputum
In a previous study, we have sorted sputum cell subsets using flow cytometry and detected the expression of miR-142-3p in sputum macrophages and neutrophils.36 To investigate the cell subsets responsible for the elevation of exosomal miR-142-3p in IPF sputum, we performed correlation analysis between miR expression levels and sputum cell subsets, particularly sputum macrophages and neutrophils (figure 2). This analysis revealed that the level of exosomal miR-142-3p was positively associated with the percentage of sputum macrophages (Spearman r=0.576, p=0.012) (figure 2A) and was negatively associated with the percentage of sputum neutrophils (Spearman r=−0.611, p=0.007) of patients with IPF (figure 2D), suggesting that macrophages are responsible for the elevation of exosomal miR-142-3p in IPF sputum. In addition, there was a positive correlation between exosomal Let-7d-5p and the percentage of sputum neutrophils (Spearman r=0.522, p=0.033) (figure 2F). There is no correlation between miR expression levels and lymphocytes or eosinophils from IPF sputum (online supplementary figure S1). In a physiological context, we did not observe any correlation between exosomal miR expression levels and sputum macrophages or neutrophils (online supplementary figure S2). Taken together, these results suggest that macrophages are responsible for the high level of sputum exosomal miR-142-3p in the IPF context.

Exosomal miR-142-3p is positively associated with sputum macrophages in IPF context. (A–C) Spearman correlation between the percentage of sputum macrophages and expression level of (A) exosomal miR-142-3p, (B) exosomal miR-33a-5p and (C) exosomal Let-7d-5p from sputum of patients with IPF. (D–F) Spearman correlation between the percentage of sputum neutrophils and expression level of (D) exosomal miR-142-3p, (E) exosomal miR-33a-5p and (F) exosomal Let-7d-5p from sputum of patients with IPF. (A–F) Data are non-normally distributed and are analysed using Spearman correlation. IPF, idiopathic pulmonary fibrosis; miR, microRNA.
Biological pathways associated with IPF-related MiRs
In order to elucidate the potential role of IPF-related miRs (miR-142-3p, miR-33a-5p and Let-7d-5p), we subjected them to in silico analysis with mirPath V.3 using Tarbase V.7 database. The analysis revealed that these miRs were involved in the regulation of transforming growth factor β (TGF-β) signalling pathway (false discovery rate (FDR)=1.45E-6) (figure 3A), a major process involved in the initiation and progression of pulmonary fibrosis by inducing epithelial cell/fibroblast differentiation, migration, invasion and deposition of extracellular matrix (ECM).5 Among the direct targets belonging to the TGF-β signalling pathway, the receptor transforming growth factor β receptor 1 (TGFβ-R1) was identified as a common target for the three IPF-related miRs (online supplementary figure S3).

miR-142-3p represses profibrotic response to TGF-β1 by targeting TGFβ-R1 in alveolar epithelial cells and lung fibroblasts. (A) In silico analysis to predict functional role of IPF-related miRs (miR-142-3p, miR-33a-5p and Let7d-5p) with mirPath v.3 (Tarbase V.7 database). (B–D) MiR-142-3p overexpression suppresses the induction of TGFβ-R1 mRNA in (B) alveolar epithelial cell line (A549) (n=5), (C) lung fibroblast cell line (MRC5) (n=3) and (D) primary lung fibroblasts (HLF) (n=5), as assessed by qRT-PCR. Cells were either unstimulated or treated with TGF-β1 (5 ng/mL) for 4 hours. Data passed the Shapiro-Wilk normality test and are expressed as mean (±SEM) and analysed using paired two-tailed t-test. (E–H) Western blotting analysis confirms that TGFβ-R1 protein is suppressed by the miR-142-3p overexpression in alveolar epithelial cell line (A549) (E,F) and lung fibroblast cell line (MRC5) (G,H). (I–K) MiR-142-3p overexpression reduces the induction of profibrotic genes (COL1A1 and TGF-β1) in response to TGF-β1 stimulation in the (I) alveolar epithelial cell line (A549) (n=5), (J) lung fibroblast cell line (MRC5) (n=3) and (K) primary lung fibroblasts (HLF) (n=5), as assessed by qRT-PCR. Data passed the Shapiro-Wilk normality test and are expressed as mean (±SEM) and analysed using paired two-tailed t-test. FDR, false discovery rate; HLF, human lung fibroblast; IPF, idiopathic pulmonary fibrosis; miR, microRNA; mRNA, messenger RNA; qRT-PCR, quantitative reverse transcription–PCR; TGF-β1, transforming growth factor β1; TGFβ-R1, transforming growth factor β receptor 1.
miR-142-3P represses TGF-β-induced fibrotic response in alveolar epithelial cells and lung fibroblasts by targeting TGFβ-R1
We next sought to investigate the biological effect of IPF-related miRs on fibrotic response. For this, miR-142-3p and miR-33a-5p were overexpressed by mimic transfection in A549 alveolar epithelial cells, and MRC5 lung fibroblasts and their impact on the expression of TGFβ-R1 were assessed. The overexpression of miR-142-3p reduces the expression of TGFβ-R1 mRNA in A549 and MRC5 cells (figure 3B,C). To validate our findings, we also confirmed the regulation in primary lung fibroblasts (human lung fibroblast (HLF)) (figure 3D). Then, we confirmed the reduction of TGFβ-R1 at protein level in A549 and MRC5 cells by miR-142-3p mimic (figure 3E–H). The overexpression of miR-33a-5p in A549 and MRC5 cells did not affect the expression of the TGFβ-R1 transcript (online supplementary figure S4).
As miR-142-3p reduced the expression of TGFβ-R1, a key element of TGF-β signalling, we anticipated that this miR would display antifibrotic properties. Indeed, our results showed that miR-142-3p suppressed the induction of the profibrotic genes by TGF-β stimulation in A549 alveolar epithelial cells (COL1A1, p=0.001; COL3A1, p=0.001; and TGF-β1, p=0.006) (figure 3I) and in MRC5 lung fibroblasts (COL1A1, p=0.006, and TGF-β1, p=0.017) (figure 3J). The antifibrotic effect of miR-142–3 p was also observed in primary lung fibroblasts (HLF) (suppression of the expression of COL1A1, p=0.041, and TGF-β1, p=0.007) (figure 3K).
We also investigated the impact of miR-142-3p on the proliferation of A549 and MRC5, as aberrant proliferation of fibroblasts plays a critical role in the pathogenesis of pulmonary fibrosis. Interestingly, mir-142-3p was able to reduce the proliferation of A549 (p=0.018) and MRC5 (p=0.048) cells (online supplementary figure S5).
These data highlight the antifibrotic property of miR-142-3p in lung epithelial cells and fibroblasts via the targeting of TGFβ-R1. In addition, miR-142-3p is able to repress lung fibroblast proliferation.
Macrophage-derived exosomes transfer miR-142-3p to recipient lung epithelial cells and fibroblasts
Because our results suggest that sputum macrophages are responsible of the elevated level of exosomal miR-142-3p in the IPF context, the next step was to study the impact of macrophage-derived exosomes on the expression of profibrotic genes. To this end, we isolated exosomes from culture medium of THP1 macrophages (figure 4A). As expected, this macrophage cell line is enriched in miR-142-3p compared with alveolar epithelial cells (A549) and lung fibroblasts (MRC5) (more than 3000-fold, p<0.001) (figure 4B). The isolated vesicles present an average size distribution of 143.4 nm (±4.4 nm) (figure 4C) and an enrichment of several exosomal markers, CD81, CD9 and synthenin, and an absence of mitochondrial cytochrome c (figure 4D), which confirms the purity of our exosome preparation.
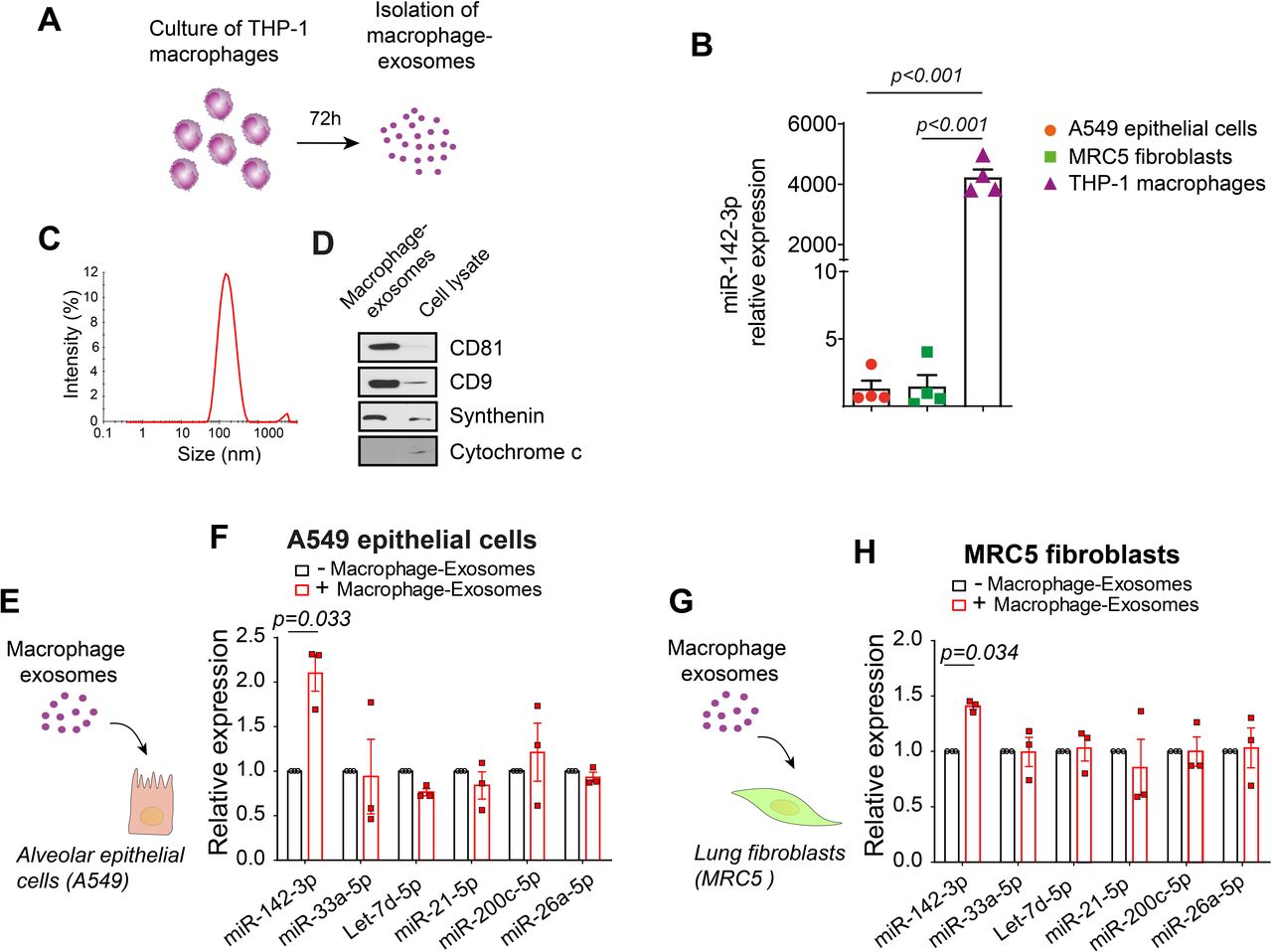
Macrophage-derived exosomes transfer miR-142-3p to recipient alveolar epithelial cells and lung fibroblasts. (A) Schematic representation of the isolation of exosomes from THP1 macrophages. Macrophages were incubated for 72 hours. Then, conditioned medium was collected and macrophage-derived exosomes were isolated using standard ultracentrifugation protocol. (B) miR-142-3p is highly enriched in THP1 macrophages compared with A549 lung epithelial cells and MRC5 fibroblasts (n=4). (C) Representative size distribution of purified THP1 macrophage-derived exosomes by dynamic light scattering analysis. A representative experiment of 3 is shown. The average size distribution of the vesicles is 143.4±4.4 nm. (D) Western blot analysis of exosomal markers, CD81, CD9, synthenin and mitochondrial protein cytochrome c in THP1 macrophage-derived exosomes and cell lysates from THP1 cells. (E,G) Schematic representation of the treatment of A549 alveolar epithelial cells (E) and MRC5 lung fibroblasts (G) with macrophage-derived exosomes for 24 hours. (F,H) Transcript levels of IPF-related miRs (miR-142-5p, miR-33a-5p, Let-7d-5p, miR-21-5p, miR-200c-5p and miR-26a-5p) assessed by qRT-PCR in A549 alveolar epithelial cells (F) and MRC5 lung fibroblasts (H) treated with macrophages-derived exosomes (5 µg) for 24 hours (n=3). (B,F,H) Data passed the Shapiro-Wilk normality test and are expressed as mean (±SEM) and analysed using paired two-tailed t-test. IPF, idiopathic pulmonary fibrosis; miR,microRNA.
Then, we tested whether miR-142-3p could be transferred to alveolar epithelial cells and lung fibroblasts via macrophage-derived exosomes. For this, we incubated A549 and MRC5 cells with THP1 macrophage exosomes for 24 hours and measured the expression of IPF-related miRs (figure 4E,G). The level of miR-142-3p increased in A549 and MRC5 cells in the presence of macrophage-derived exosomes (2.1 fold, p=0.033, and 1.41 fold, p=0.034, respectively) (figure 4F,H), demonstrating that these vesicles are able to transfer miR-142-3p to target cells.
Macrophage-derived exosomes are able to suppress profibrotic activation in lung epithelial cells and fibroblasts
Because macrophage-derived exosomes are able to transfer antibrotic miR-142-3p to target cells, we anticipated that these extracellular vesicles (EVs) will present antifibrotic properties in the recipient cells. To test this, we incubated A549 and MRC5 cells with purified THP1 macrophage-derived exosomes for 24 hours and treated the cells with TGF-β for 4 hours (figure 5A,B). Macrophage-derived exosomes were able to reduce TGFβ-R1 transcript in A549 epithelial cells (p=0.013) and MRC5 fibroblasts (p=0.013) (figure 5C,F) and its protein level in A549 cells (figure 5D,E). Furthermore, they were able to repress the expression of profibrotic genes in the presence of TGF-β in A549 (COL1A1, p=0.032; COL3A1, p=0.002; and TGF-β1, p=0.003) and MRC5 cells (COL1A1, p=0.015; COL3A1, p=0.038; and TGF-β1, p=0.022) (figure 5G,H).

Macrophage-derived exosomes present antifibrotic property. (A,B) Schematic representation of the treatment of A549 lung epithelial cells (A) and MRC5 fibroblasts (B) with macrophage-derived exosomes for 24 hours, followed by TGF-β1 stimulation (4 hours). (C–E) Macrophage-derived exosomes repress TGFβ-R1 expression at transcript (C) (n=4) and protein (D,E) levels in A549 lung epithelial cells. (F) Macrophage-derived exosomes repress the expression of TGFβ-R1 mRNA in MRC5 lung fibroblasts (n=4). (G,H) Macrophage-derived exosomes repress the induction of profibrotic genes (COL1A1, COL3A1 and TGF-β1) in response to TGF-β1 stimulation in (G) the alveolar epithelial cell line (A549) (n=4) and (H) the lung fibroblast cell line (MRC5) (n=4), as assessed by qRT-PCR. (C,F,G,H) Data passed the Shapiro-Wilk normality test and are expressed as mean (±SEM) and analysed using paired two-tailed t-test. qRT-PCR, quantitative reverse transcription PCR; TGF-β1, transforming growth factor β1; TGFβ-R1, transforming growth factor β receptor 1.
In order to demonstrate that miR-142-3p is the active element responsible for the antifibrotic property of macrophage-derived exosomes, miR-142-3p was knocked down in THP1 macrophages prior to exosome collection and treatment of A549 cells (figure 6A,B). This affects the antifibrotic property of corresponding exosomes. Indeed, we observed an upregulation of the expression of profibrotic genes in alveolar epithelial cells treated with exosomes derived from miR-142-3p inhibitor-treated macrophages compared with the ones derived from control inhibitor-treated macrophages (COL1A1, p=0.045; COL3A1, p=0.007; and TGF-β1, p=0.018) (figure 6D–F).

Antifibrotic property of macrophage-derived exosomes is miR-142-3p dependent. (A) Schematic representation of the treatment of A549 lung epithelial cells with miR-142-3p depleted macrophage exosomes. The level of miR-142-3p was silenced in THP1 macrophages by transfection of the miR-142-3p inhibitor. After 24 hours, the culture medium was refreshed and miR-142-3p depleted macrophage exosomes were isolated after additional 72 hours. Then, their impact on the modulation of the expression of profibrotic genes in A549 lung epithelial cells in the presence of TGF-β1 was evaluated. (B) The level of miR-142-3p was reduced in A549 cells after 24 hours of miR-142-3p inhibitor transfection. (C–F) Inhibiting miR-142-3p in THP1 macrophages affects the antifibrotic property of related exosomes, as shown by their impact on the expression of TGFβ-R1 (C), COL1A1 (D), COL3A1 (E) and TGF-β1 (F), compared to crtl inhibitor-exosomes. (B–F) Data passed the Shapiro-Wilk normality test and are expressed as mean (±SEM) and analysed using paired two-tailed t-test. miR, microRNA; mRNA, messenger RNA; PBS, phosphate-buffered saline; TGF-β1, transforming growth factor β1; TGFβ-R1, transforming growth factor β receptor 1.
Taken together, these findings delineate a process whereby transfer of miR-142-3p from macrophage-derived exosomes to alveolar epithelial cells and lung fibroblasts can modulate profibrotic gene expression and thereby could attenuate pulmonary fibrosis.
Discussion
Exosomes have emerged as essential actors impacting the progression of fibrotic diseases by transferring antifibrotic or profibrotic miRs to target cells and affecting pathological fibrogenesis. The objective of our study was to elucidate the impact of IPF-related exosomal miRs on fibrogenesis. Here, we found that miR-142-3p is upregulated in sputum and plasma of patients with IPF, suggesting that this miR influences the pathogenesis of pulmonary fibrosis. Correlation analysis revealed a positive association between exosomal miR-142-3p and the percentage of macrophages from the sputum of patients with IPF, suggesting macrophage origin of miR-142-3p upregulation. In accordance with our findings, previous studies have shown that primary macrophages, including sputum macrophages, express high amount of miR-142-3p.36 37 In addition, Squadrito et al found that macrophages are able to release exosomes containing miR-142-3p, which can be transferred to target cells.37 An elegant study from Maes et al has shown that miR-142-3p was significantly upregulated in sputum of patients with severe asthma.36 Interestingly, a positive correlation between miR-142-3p and sputum neutrophils was observed in asthmatic context. Depending on pathological context, it appears that upregulation of sputum miR-142-3p derives from macrophages or neutrophils, two sputum cell subsets expressing miR-142-3p. In the study from Maes et al, the detection of miR-142-3p was performed on sputum supernatants, so the miR could be either present in the EV fraction, which contains exosomes and microvesicles and/or in the EV-free fraction. It would be interesting to isolate exosomes from asthma sputum in order to investigate the expression of exosomal miR-142-3p in the asthmatic context.
In silico analysis and functional experiments on alveolar epithelial cells and lung fibroblasts transfected with mimics indicated that miR-142-3p presents antifibrotic properties by modulating TGF-β signalling pathway via targeting the receptor TGFβ-R1. The major cellular sources of TGF-β production during IPF have been identified as alveolar macrophages, airway epithelial cells and lung fibroblasts.38–40 We thus favour the hypothesis that miR-142-3p could slow the progression of IPF by blocking the production of TGF-β in airway epithelial cells and lung fibroblasts. In addition, we have shown that miR-142-3p represses the expression of ECM-related genes (COL1A1 and COL3A1) which could reduce the deposition of ECM and scar formation. In accordance with our findings, Wang et al demonstrated that overexpression of miR-142-3p represses the expression of profibrotic genes in cardiomyocytes by targeting high mobility group box 1.41 Likewise, Yang et al reported that the plasma level of miR-142-3p declined significantly in patients with hepatic cirrhosis, and ectopic expression of miR-142-3p in activated hepatic stellate cells decreased profibrotic markers.42
Chronic injury of lung tissue is an early event in IPF which leads to an irreversible alteration of the reparative functions of alveolar epithelial cells.43 One of the key factors responsible for the alteration of reparative capacity of alveolar epithelial cells is senescence.44 The inhibition of TGF-β/TGFβ-R1 complex by exosomal miR-142-3p transfer may prevent TGFβ-induced senescence in these cells and preserve their self-renewal capacity. In addition, direct repression on senescence molecular pathways via miR-142-3p could also contribute alveolar repair since some key players such as p53 are direct targets of miR-142-3p.45 Another study by Guo et al has shown that miR-142-3p could modulate pulmonary fibrosis by inhibiting apoptosis and inflammation induced by bleomycin through down-regulation of cyclooxygenase-2.46 Recent studies have demonstrated the important contribution of mitochondrial dysfunction in initiation and maintenance of pathological fibrogenesis.47 48 Future investigations will be needed to assess the impact of miR-142-3p on alveolar epithelial cell renewal capacity and endoplasmic reticulum stress.
We also showed that miR-142-3p partially represses the proliferation of lung fibroblast, a process playing a critical role in the pathogenesis of lung fibrosis. In accordance with our findings, Carraro et al demonstrated that miR-142-3p contributes to the proper proliferation of mesenchymal progenitors during lung development by controlling the level of wingless-related integration site (WNT) signalling.49 In addition, recent studies have reported the antiproliferative property of miR-142-3p in tumourous context.50 51
One of the major questions in the field of EVs is the role of exosomes in pathological contexts, particularly lung fibrosis. Several studies have reported that exosomes play versatile roles, depending on the disease and their cell origin. Some studies have demonstrated that mesenchymal stem cell-derived exosomes are able to facilitate wound healing and blocks myofibroblastic differentiation in IPF context,52 53 whereas others have suggested deleterious effects.54 55 Those discrepancies are probably due to distinct cells and tissue origin of those vesicles. Here, we found that macrophage-derived exosomes present antifibrotic properties in vitro due in part to the transfer of miR-142-3p to alveolar epithelial cells and lung fibroblasts. The upregulation of antifibrotic miR-142-3p in IPF context is probably a process to fight against fibrogenesis. Indeed, several studies as demonstrated that specific miRs are upregulated in pathological context and act as mediators of negative feedback loop to turn down pathological processes.56 Our result suggest that macrophage-derived exosomes may fight against pulmonary fibrosis progression via the delivery of antifibrotic miR-142-3p. To our knowledge, this is the first study mentioning the protective role of macrophage-derived exosomes in IPF context. A perspective of our work is the use of miR-142-3p-loaded exosomes as potential therapeutic tool for the treatment of lung fibrosis. Further investigations need to be performed on a mouse model of pulmonary fibrosis (eg, bleomycin model).
A limitation of our study is the use of two independent cohorts to study sputum-derived exosomes and plasma-derived exosomes. Furthermore, the size sample of both cohorts are small (sputum cohort (19 IPF vs 23 HS) and plasma cohort (14 IPF vs 14 HS)) and heterogeneous. It is due to the fact that IPF is a rare disease, which makes it difficult to generate cohorts. The relatively low number of studied samples of the correlation analysis may have contributed to borderline p values. Finally, we were unable to obtain IPF sputum in order to isolate sputum cell subsets to assess the levels of miR-142-3p. However, in one of our previous studies, we have already detected the expression of miR-142-3p in sputum macrophages and neutrophils.36
In summary, our study provides a new finding for the role of macrophage-derived exosomes in modulating fibrosis in pulmonary fibrosis diseases. We propose that the antifibrotic properties of macrophage-derived exosomes due in part to the delivery of miR-142-3p to target cells may slow the progression of pulmonary fibrosis by targeting excessive deposition of ECM (figure 7). The delivery of this antifibrotic miR in the lung could protect against fibrosis, revealing a new therapeutic potential to treat IPF disease.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed model illustrating the antifibrotic property of macrophage-derived exosomes in the idiopathic pulmonary fibrosis context. Macrophage-derived exosomes are able to slow the progression of pulmonary fibrosis by targeting excessive deposition of ECM and aberrant proliferation of lung fibroblasts via the delivery of antifibrotic miR-142-3p. ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition; miR, microRNA; TGF-β, transforming growth factor β.
References
Footnotes
IS and M-SN contributed equally.
Contributors JG designed and performed the experiments, analysed the data and wrote the manuscript; MC, AB and ON performed the experiments and analysed the data; MH collected and processed sputum and plasma samples; FG, EL, MM, FD and RL revised the manuscript; IS and M-SN designed, analysed and supervised the experiments, and wrote and revised the manuscript. All authors read and approved the final manuscript.
Funding This study was supported by the Fonds d’Investissement de Recherche Scientifique du Centre Hospitalier Universitaire de Liège, Centre Hospitalier Universitaire de Liège, the University of Liège, the Fonds National de la Recherche Scientifique and the fonds Léon Frédéricq.
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval The protocol was approved by the Ethics Committee of CHU of Liège, and all subjects gave written consent before their enrolment (Belgian number: B707201422832, ref: 2014/302).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.
Linked Articles
- Airwaves