Article Text

Abstract
Background Respiratory syncytial virus (RSV) is associated with childhood asthma. Nevertheless, not all children exposed to RSV develop asthma symptoms, possibly because genes modulate the effects of RSV on asthma exacerbations.
Objective The purpose of this study was to identify genes that modulate the effect of RSV latent infection on asthma exacerbations.
Methods We performed a meta-analysis to investigate differentially expressed genes (DEGs) of RSV infection from Gene Expression Omnibus datasets. Expression quantitative trait loci (eQTL) methods were applied to select single nucleotide polymorphisms (SNPs) that were associated with DEGs. Gene-based analysis was used to identify SNPs that were significantly associated with asthma exacerbations in the Taiwanese Consortium of Childhood Asthma Study (TCCAS), and validation was attempted in an independent cohort, the Childhood Asthma Management Program (CAMP). Gene–RSV interaction analyses were performed to investigate the association between the interaction of SNPs and RSV latent infection on asthma exacerbations.
Results A total of 352 significant DEGs were found by meta-analysis of RSV-related genes. We used 38 123 SNPs related to DEGs to investigate the genetic main effects on asthma exacerbations. We found that eight RSV-related genes (GADD45A, GYPB, MS4A3, NFE2, RNASE3, EPB41L3, CEACAM6 and CEACAM3) were significantly associated with asthma exacerbations in TCCAS and also validated in CAMP. In TCCAS, rs7251960 (CEACAM3) significantly modulated the effect of RSV latent infection on asthma exacerbations (false-discovery rate <0.05). The rs7251960 variant was associated with CEACAM3 mRNA expression in lung tissue (p for trend=1.2×10−7). CEACAM3 mRNA was reduced in nasal mucosa from subjects with asthma exacerbations in two independent datasets.
Conclusions rs7251960 is an eQTL for CEACAM3, and CEACAM3 mRNA expression is reduced in subjects experiencing asthma exacerbations. CEACAM3 may be a modulator of RSV latent infection on asthma exacerbations.
- asthma genetics
- paediatric asthma
- viral infection
- asthma pharmacology
- clinical epidemiology
Statistics from Altmetric.com
Key messages
What is the key question?
Do genetic variants modulate the effects of respiratory syncytial virus (RSV) latent infection on asthma exacerbations in children?
What is the bottom line?
We found that rs7251960 (CEACAM3) modifies the effects of RSV latent infection on asthma exacerbations. We also identified eight RSV-related genes (GADD45A, GYPB, MS4A3, NFE2, RNASE3, EPB41L3, CEACAM6 and CEACAM3) that are significantly associated with asthma exacerbations.
Why read on?
To our knowledge, this is the first study to examine the interaction of gene–RSV latent infection on asthma exacerbations on a transcriptome-wide scale. Linking gene expression profiles to a gene–RSV interaction model may provide biological insight on the pathogenesis and treatment of asthma exacerbations.
Introduction
Asthma is the most common chronic disease among children,1 and respiratory tract infections can trigger asthma symptoms.2 3 Respiratory syncytial virus (RSV) has been recognised as a major cause of respiratory tract infections4–6 and is associated with asthma exacerbations and failure to respond to asthma medications.7–10 Nocturnal wheezing and speech-limiting severe wheezing are indicators of asthma exacerbations, which require short-acting beta-agonists (eg, albuterol), oral corticosteroid bursts, emergency department visits or hospitalisations.11 12 Although severe RSV infection during early life is associated with wheezing and asthma in later childhood,13–15 little is known about the effects of the risk of asthma exacerbations when infected by RSV. Host genetics may explain why some children experience severe asthma exacerbations in the setting of RSV infection, while others do not.
Studies have examined changes in gene expression with respect to the pathophysiology of RSV infection,16–21 and Brand et al 17 reported that OLFM4 expression is significantly increased during the acute phase of RSV infection compared with the recovery phase. Other studies demonstrated that interferon-related genes are overexpressed in children infected with RSV16 18; however, these studies focused on healthy populations and not individuals with asthma. Furthermore, some genes associated with RSV infection, including IL-10, TGFB1 and ADAM33, are linked to an increased risk of asthma.15 Nevertheless, the aforementioned studies used candidate gene approaches. Although researchers can gain insight from investigating the interaction of candidate genes and exposure to RSV, novel genetic variants might be missed. Most genome-wide gene–environment interaction studies are underpowered to detect significant genetic variants and lack information on the mechanisms involved.22 Therefore, assessing genome-wide gene expression profiles in response to viruses of interest to select potential gene targets may help elucidate gene–environment interactions. To the best of our knowledge, no studies have used a two-step integrative approach on a transcriptome-wide scale to examine the role of gene–RSV interactions on asthma exacerbations.
The objective of this study was to identify SNPs that modulate the effect of RSV infection on asthma exacerbations on a transcriptome-wide scale. We also aimed to investigate the associations between SNPs, genes and asthma exacerbations in multiple populations. Linking gene expression profiles to a gene–RSV interaction model may provide biological insight on the nature of asthma exacerbations.
Materials and methods
Target genes and SNPs selection
Meta-analysis of RSV-related genes
Five gene expression studies that included RSV-related genes were identified through Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) using the search terms ‘RSV’ and ‘blood’ (online supplementary). The RankProd method23 24 from OMics Compendia Commons (OMiCC, https://omicc.niaid.nih.gov/) was used to identify differentially expressed genes (DEGs) by integrating multiple array platforms to detect genes that were consistently ranked highly in multiple studies. Briefly, we created comparison group pairs (CGPs) where each CGP comprises two conditions of gene-expression profiles (eg, RSV infection vs healthy controls) in each study. After CGPs were defined, the quantile normalisation and Limma25 procedures were used to calculate expression differences between RSV infected and healthy controls in each study. Meta-analysis was performed using the RankProd23 24 method to convert fold-change value into ranks within a CGP. Then, a rank product was calculated for each gene across the CGPs. Lastly, p value and false-discovery rate (FDR) were calculated based on permuted expression value. DEGs were considered significant if they had a FDR less than 0.05 to account for multiple testing. Additionally, we visualised gene expression values across studies by using hierarchical clustering with heatmap.2 function of gplots.26 Normalised gene expression values were calculated with the Combat module from sva package27 to adjust for studies with varying gene expression values.
Supplemental material
Expression quantitative trait loci (eQTL) analysis and SNP identification
We used whole blood datasets from Genotype-Tissue Expression (GTEx, V6p release) to find genetic variants related to gene expression. The detailed procedure for the eQTL analysis is found on the GTEx website (https://www.gtexportal.org/home/). Cis-regulating SNPs were defined as the SNPs within a region bounded by 106 bp distance from both ends of the transcrition start site of each gene. Significant SNP–gene pairs (cis-eQTLs, FDR<0.05) from GTEx were included. SNPs located in the gene range ±5 kb were also included in the present study because we considered SNPs within transcription start sites, promoter regions, exons and introns to potentially regulate gene expression.28
Discovery population
The discovery population was the Taiwanese Consortium of Childhood Asthma Study (TCCAS), a consortium-based study for childhood asthma, comprising of several paediatric study populations in Taiwan. We recruited children with asthma from outpatient paediatric clinics. Inclusion criteria included asthma diagnosed by an asthma specialist and onset of asthma at age less than 10 years. To minimise the heterogeneity of the study population, exclusion criteria included cancer; major immunological diseases, such as systemic lupus erythematosus, Henoch-Schonlein purpura; rare hereditary diseases, such as type I diabetes or Marfan syndrome; or severe infection.
DNA preparation and genotyping
DNA was extracted from peripheral blood using the QIAamp DNA mini kit (QIAGEN Inc, Catalog No. 51104). Whole-genome genotyping was performed using the Affymetrix Axiom Genome-Wide CHB 1 Array Plate. Genotype imputation was performed using IMPUTE2,29 and individuals from phase 3 of the 1000 Genomes Project were used as reference. We excluded the imputed SNPs with R2 values <0.3. Markers were excluded if minor allele frequencies were less than 0.01, or call rates were less than 95%. We removed one individual from each pair with an identity by descent (IBD) >0.1875, which is halfway between the expected IBD for third-degree and second-degree relatives.30 The total genotyping rate in the remaining individuals was 0.99.
RSV latent infection measurement
We used a commercial ELISA kit to assess the units of IgM antibodies (Arigo, ARG80601) to RSV following the manufacturer’s protocol. In order to eliminate interference from hyperimmune levels of IgG, sera were pretreated with anti-IgG. Samples containing more than 12 units of IgM were considered positive, as suggested by the manufacturer’s instructions.31 In addition to RSV infection, we measured RSV-specific IgE to distinguish subjects with and without sensitisation to RSV (online supplementary). An optical density reading of two SDs above the average control values was defined as positive sensitisation to RSV.32 Samples from 46 non-asthmatic controls were measured to calculate the cut-off point, and the mean was 0.22 and SD was 0.18.
Definition of asthma outcomes
We collected information on asthma outcomes by interview from children and their parents or guardians at the time of recruitment in TCCAS. An asthma exacerbation was defined as answering ‘yes’ to one of the following questions: ‘Do you (Does this child) ever have any nocturnal wheezing and cough during the past 2 weeks?’ or ‘Do you (Does this child) ever have any speech-limiting severe wheezing and shortness of breath during the past 2 weeks?’33 Pulmonary function tests were performed by fully trained technicians using a spirometer (Chestgraph HI-101, CHEST M.I., INC). Each subject performed three satisfactory exhalation manoeuvres in which the two highest values for the FVC values and the two highest FEV1 values were consistent within 150 mL, extrapolation volume was less than 150 mL or 5% of FVC and forced expiratory time exceeded 6 s. These criteria were based on American Thoracic Society and European Respiratory Society recommendations, updated in 2005 and modified for children.34
Validation population
The Childhood Asthma Management Program (CAMP) was a clinical trial that followed 1014 children ages 5–12 years with mild-to-moderate asthma for 4 years.35 Participants provided DNA for genetic analysis, and informed consent was obtained. Clinical biomarkers were measured at baseline; spirometry, bronchial hyper-responsiveness and asthma exacerbations were recorded both at baseline and at each follow-up visit. In this study, we included 833 subjects who had whole-genome genotyping data and asthma exacerbation outcomes. Genome-wide SNP genotyping was performed using the HumanHap550v3 Genotyping BeadChip (Illumina). Imputation was performed on the basis of 1000 Genomes Project as previously described.36 The children or their parents or guardians completed diary cards that recorded asthma outcomes each day.35 Diary cards were included in the analysis if the subject completed at least 24 days of entries for a 28-day period. In CAMP, asthma exacerbations were defined as answering ‘yes’ to one of the following questions between study recruitment and the 2-month follow-up visit: ‘wheezing-induced nocturnal awakenings at least once a month’ and ‘limitation of activity due to asthma at least once a month’.
Statistical analyses
We used logistic regression with both dominant and additive genetic models to investigate the effects of the SNPs on asthma exacerbations. Models were adjusted for age, sex, asthma medication use, body mass index (BMI) and the first two principal components in TCCAS. Asthma medications studied included inhaled corticosteroid with or without a long-acting β2-agonist used within 2 weeks of the time of recruitment. We used gene-based analysis to identify SNPs that were significantly associated with asthma exacerbations, and empirical p values were calculated with 1000 permutations using the set-test method in PLINK.37 Statistics from a set of SNPs (single association p value <0.05) within or close to a gene were aggregated. Only SNPs having r2 <0.5 with each other were retained for each gene to account for the linkage disequilibrium (LD) among SNPs. Validation was performed in CAMP if the SNP association in TCCAS showed a permutation p value of less than 0.05. Fisher combined p values were calculated using the sum of logs method from two cohorts with the same direction of effects. To account for multiple comparisons, FDR adjustments were fitted to estimate the effects of SNPs on asthma exacerbations. Likelihood ratio tests were used to detect the interaction between SNPs and RSV latent infection on asthma exacerbations in TCCAS, and FDR adjustments were also used for multiple comparisons. The interaction model was specified as follows: asthma exacerbation=SNP (dominant or additive model)+RSV latent infection (no/yes)+SNP*RSV latent infection+covariates. Covariates included age, sex, asthma medication use, BMI, RSV sensitisation and the first two principal components. Analyses were completed with the software packages PLINK 1.9 (https://www.cog-genomics.org/plink/1.9)38 and RStudio.39
eQTL, mRNA analyses and ENCODE analysis of target SNPs
We investigated the target SNPs for association with mRNA levels in lung tissue by using the results of the eQTL analysis from the GTEx database and the TCCAS cohort. To determine the expression of CEACAM3 in nasal epithelial samples from subjects with asthma, we used two gene expression datasets from the GEO resource. The GSE1919040 dataset includes Affymetrix Human Gene 1.0 ST Array data for six uncontrolled and seven controlled asthma subjects. We also used the GSE4617141 dataset, which includes Agilent Whole Human Genome 4×44K, for 10 asthmatics with exacerbations and 24 asthmatics without exacerbations. To provide insight on the functional regulation of SNPs and mechanism of disease, we investigated significant SNPs using the HaploReg42 and the deep-learning functional prediction resource DeepSEA43 (online supplementary).
Results
The results are summarised in figure 1. From GEO, we found five RSV genome-wide gene expression datasets with seven gene lists (table 1). Meta-analysis identified 352 DEGs after FDR correction (FDR<0.05), of which 323 were upregulated and 29 were downregulated with a fold change >1.5 (online supplementary figure S1). A full list of the 352 DEGs, sorted by mean log2 fold change are shown in online supplementary table S1 and S2. The heatmap of the 352 DEGs across studies is shown in figure 2.
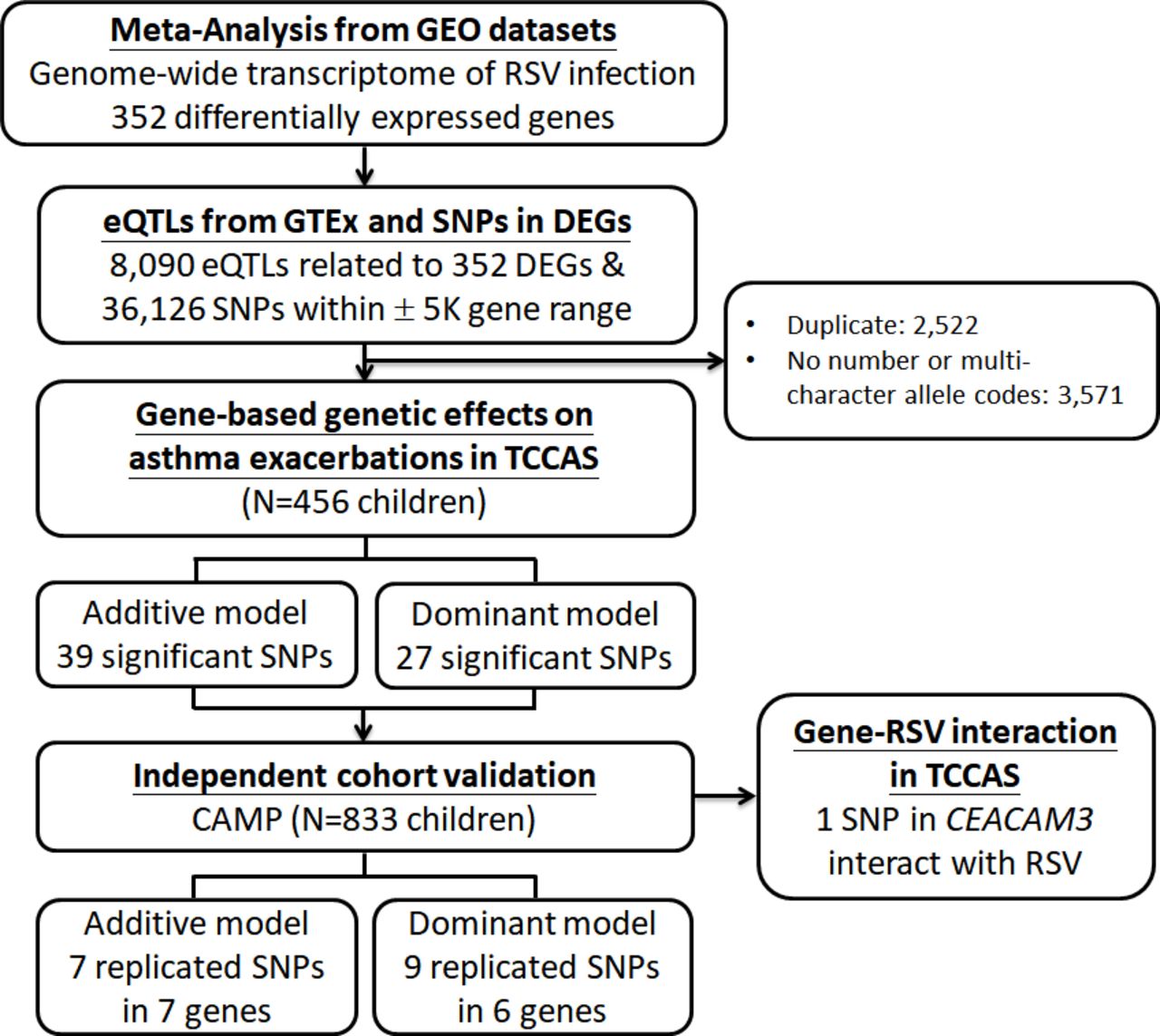
Flow chart showing study design and results. CAMP, Childhood Asthma Management Program; CEACAM3, carcinoembryonic antigen-related cell adhesion molecules 3; DEGs, differentially expressed genes; eQTL, expression quantitative trait loci; GEO, Gene Expression Omnibus; GTEx, Genotype-Tissue Expression; RSV, respiratory syncytial virus; TCCAS, Taiwanese Consortium of Childhood Asthma Study.
Genome-wide gene expression studies from human peripheral blood related to RSV infection

Differences in gene expression between RSV infection acute phase and recovery phase/healthy controls. A total of 352 differentially expressed genes with FDR <0.05 and a >1.5 fold change was arranged using hierarchical clustering with heatmap.2 function of gplots26 in R. Each row represents a normalised gene expression value, and each column represents a subject. Colours represent normalised gene expression value in each subject, with blue indicating high expression and yellow indicating low expression. Normalised gene expression values were calculated with the Combat module from sva package27 in R to adjusted for different studies with varying gene expression values. Each gene list is divided by a vertical black line. GSE69606_s: severe cases; GSE69606_m: moderate cases. FDR, false-discovery rate; RSV, respiratory syncytial virus.
We extracted eQTLs from whole blood in the GTEx dataset, and 8090 cis-eQTLs were associated with 352 RSV-related genes. In order to increase the discovery of SNPs related to RSV, SNPs within a region of 5 kb of the gene were also included. We investigated the genetic effects of a total of 38 123 RSV-related SNPs on asthma exacerbations. Finally, we studied the validated SNPs in the gene–RSV interaction analysis. The summary of RSV-related genes and selection of SNPs is shown in figure 1.
A total of 456 subjects with asthma were analysed in TCCAS. The mean age in TCCAS was 10.5±3.4 years, and 68% were male (table 2). The mean age of CAMP was 9.0±2.1 years, and 61% were male. The subjects had a wide range of asthma severity, from mild to severe, according to the GINA classification. While all of the subjects in the TCCAS population were Han Chinese, subjects in CAMP were of multiple races. The rate of positive RSV-specific IgM was 7.5% (34/456) in TCCAS. In subjects with RSV latent infection, 44% experienced asthma exacerbations and in subjects without RSV latent infection, 35% experienced asthma exacerbations. RSV latent infection was not associated with SNPs in TCCAS (data not shown).
Clinical characteristics of children in TCCAS and CAMP
Using gene-based analysis, we identified 39 significant SNPs using an additive model and 27 significant SNPs using a dominant model (online supplementary table S3). table 3 shows the results of the association of SNPs with asthma exacerbations in TCCAS and validation in CAMP. Using an additive genetic model, a total of 39 SNPs (14 genes) were significantly associated with an increased risk of asthma exacerbations using the gene-based analysis in TCCAS (permutation p<0.05). We evaluated the 39 SNPs with the outcome of asthma exacerbations in CAMP for validation. We found that seven SNPs in seven RSV-related genes (GADD45A, GYPB, MS4A3, NFE2, EPB41L3, CEACAM6 and CEACAM3) validated in CAMP (FDR <0.05) (table 3). To further assess the genetic associations, we also used a logistic regression model with a single SNP method in TCCAS (online supplementary table S4). We found 894 SNPs were significantly associated with asthma exacerbations in TCCAS and 146 SNPs in 21 genes validated in CAMP. All of the seven RSV-related genes (GADD45A, GYPB, MS4A3, NFE2, EPB41L3, CEACAM6 and CEACAM3) in our gene-based analysis validated using the single SNP method. Using a dominant model with the gene-based analysis, six genes (MS4A3, NFE2, RNASE3, EPB41L3, CEACAM6 and CEACAM3) were associated with asthma exacerbations (table 3). SNPs in the same six genes were significantly associated with asthma exacerbations using the single SNP method (online supplementary table S5).
Results of analyses of genetic association and interaction of SNPs on asthma exacerbations
In TCCAS, rs7251960 (CEACAM3) modulated the effects of RSV latent infection on asthma exacerbations (p value for interaction=0.03, figure 3). More than 63% of subjects with asthma and RSV latent infection who had at least one copy of CEACAM3 rs7251960 (T allele) experienced asthma exacerbations. In contrast, of subjects with asthma and RSV latent infection who did not have a copy of the rs7251960 T allele, only 8.3% of subjects experienced asthma exacerbations. In adjusted models, subjects with RSV latent infection and rs7251960 CT/TT genotype had a fivefold increased odds of experiencing asthma exacerbations (OR, 5.45; 95% CI 2.04 to 14.59), compared with subjects without RSV latent infection or the rs7251960 T allele. In an additive model, among subjects with RSV latent infection, rs7251960 CT genotype was associated with a 6.9-fold increased odds of asthma exacerbations (OR 6.93; 95% CI 2.19 to 21.97).

Joint effects of CEACAM3 SNP genotypes and RSV infection on asthma exacerbations in TCCAS cohort. Cases: subjects with asthma exacerbations; controls: subjects without asthma exacerbations. Models adjusted by age, sex, asthma medication use, body mass index, principal components 1 and 2. L95, lower 95% CI of OR; RSV, respiratory syncytial virus; TCCAS, Taiwanese Consortium of Childhood Asthma Study; U95, upper 95% CI of OR.
CEACAM3 SNPs did not significantly modulate RSV latent infection status on the outcome of pulmonary function. However, with a dominant model, the trends of interactive effects for SNP-RSV infection were similar to the interactive effects on asthma exacerbations. Among subjects with RSV latent infection, the T allele in CEACAM3 (rs7251960) appeared to have potential risk effects on pulmonary function. Subjects with one copy of the CEACAM3 (rs7251960 T allele) with RSV infection had a median PEF of 3.0 L/min, while subjects without CEACAM3 rs7251960 T allele and RSV infection had a higher lung capacity (median PEF of 3.4 L/min) (online supplementary table S6). Similar effects were observed for CEACAM3–RSV interaction on FEV1. Subjects with the rs7251960 T allele and RSV infection had a median FEV1 of 1.4 L, whereas those with RSV infection but without the rs7251960 T allele had a median FEV1 of 1.8 L.
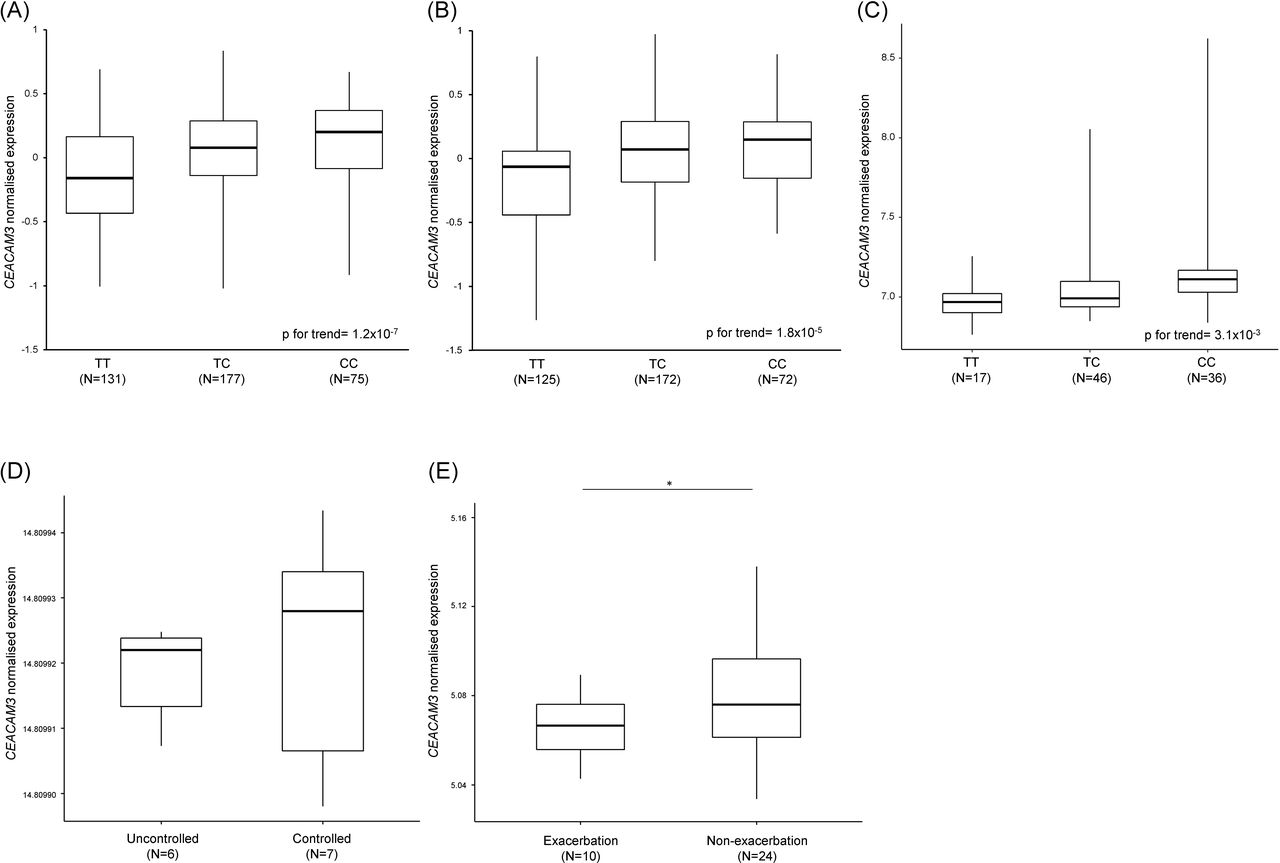
In our eQTL analyses, we found rs7251960 was an eQTL for CEACAM3 in lung tissue (figure 4A) and whole blood (figure 4B,C). The rs7251960 risk allele (T) was associated with decreased levels of CEACAM3 mRNA in lung tissue (p for trend=1.2×10−7) and in whole blood (p for trend=1.8×10−5 in GTEx; p for trend=3.1×10−3 in TCCAS). We also examined an association between CEACAM3 gene expression and asthma exacerbations in nasal mucosa samples. CEACAM3 mRNA levels were reduced in subjects with uncontrolled asthma compared with subjects with controlled asthma (figure 4D) and were reduced in subjects with asthma exacerbations compared with those without asthma exacerbations (figure 4E).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
rs7251960 is an eQTL for CEACAM3 in lung tissue and whole blood, and CEACAM3 mRNA expression is reduced in the nasal mucosa of subjects with asthma exacerbations. CEACAM3 mRNA expression is stratified by rs7251960 genotypes. (A) Lung tissue samples (n=383) are collected from GTEx. Whole blood samples are collected from (B) GTEx (n=369) and (C) TCCAS (n=99). The boxes show the median and IQR, and the whiskers show the maximum and minimum for each genotype. Trimmed mean of M-values normalised expression values were normalised across samples using an inverse normal transformation in GTEx. In TCCAS, raw gene expression levels were applied by variance stabilising transformation and robust spline normalisation. mRNA expression of CEACAM3 in (D) the GSE19190 dataset and (E) the GSE46171 dataset. Nasal epithelial samples are from uncontrolled asthma (n=6) and controlled asthma (n=7) in the GSE19190 dataset and from subjects with asthma exacerbations (n=10) and patients without asthma exacerbations (n=24) in the GSE46171 dataset. *P<0.05. eQTL, expression quantitative trait loci; GTEx, Genotype-Tissue Expression; TCCAS, Taiwanese Consortium of Childhood Asthma Study.
In the HaploReg analysis, we identified several potential functional changes of rs7251960 in CEACAM3, which altered CEBPB, COMP1, Gsc, Otx2, Pou2f2 and YY1 (online supplementary table S7). Additionally, the risk allele (T) of rs7251960 on CEACAM3 was predicted to result in a log₂ fold change of more than 0.6 in function at YY1 and Pou2f2 binding sites in various cell types in the DeepSEA analysis (online supplementary table S8).
Discussion
Our study has three key findings. First, we found that rs7251960 in CEACAM3 appears to modify the effects of RSV latent infection on asthma exacerbations. We also identified SNPs in eight RSV-related genes, GADD45A, GYPB, MS4A3, NFE2, RNASE3, EPB41L3, CEACAM6 and CEACAM3, which were significantly associated with asthma exacerbations. Furthermore, we demonstrated the utility of a novel two-step integrative approach to identify potential genes and biological pathways that may interact with RSV latent infection in asthmatic children.
Our study demonstrates that a CEACAM3 SNP interacts with RSV latent infection on asthma exacerbations in children. We also found that rs7251960 was an eQTL for CEACAM3, and CEACAM3 mRNA was reduced in nasal mucosa in subjects with uncontrolled asthma and asthma exacerbations (figure 4D, E). CEACAM3 (carcinoembryonic antigen-related cell adhesion molecules 3) is a member of the carcinoembryonic antigen (CEA) gene family and has been shown to be a granulocyte-specific CEACAM protein involved in host defence by promoting pathogen recognition and phagocytosis.44 When a respiratory tract is infected by RSV, massive granulocytes such as neutrophils are deployed to infection sites to destroy pathogens by phagocytosis. CEACAM3 is a granulocyte-specific protein, and pathogens that bind to CEACAM3 on the surface of the neutrophil activate the efficient internalisation of viruses and bacteria.45 Asthma severity has been found to correlate with increased CEACAM6 protein levels in bronchial biopsies compared with healthy controls.46 CEACAM3, expressed together with CEACAM6 and CEACAM1, has been recognised to internalise CEACAM-binding pathogens in the absence of opsonising antibodies or complement factors.44 45 Because CEACAM3 may determine the efficiency of pathogen recognition and phagocytosis as well as shift the balance towards the beneficial role of neutrophil granulocytes, it could be a potential biomarker for identifying the effect of RSV latent infection on asthma exacerbations and severity. In an analysis using ENCODE, we found that rs7251960 in CEACAM3 may be affected by alterations in YY1 and Pou2f2 transcription activity in various cell types (online supplementary table S7 and S8).47 48 We summarised the effects of CEACAM3 and associated regulatory genes of RSV and asthma in online supplementary figure S2.
Several genes, GADD45A, GYPB, MS4A3, NFE2, RNASE3 and EPB41L3, were significantly associated with asthma exacerbations in TCCAS and were validated in CAMP. GADD45A (growth arrest and DNA damage inducible alpha) encodes Gadd45a, which has been reported to modulate innate immune functions of granulocytes and macrophages.49 GYPB (glycophorin B) encodes glycophorin protein, which is a sialoglycoprotein of the human erythrocyte membrane.50 Glycophorin has been shown to be associated with decreased pulmonary function.51 Eosinophil cationic protein (ECP) is encoded by the RNASE3 (ribonuclease A family member 3) gene and is an objective measurement for asthma severity.52 Serum ECP levels in asthmatic children increase significantly during asthma exacerbations. One NFE2 (nuclear factor, erythroid 2)-related protein, Nrf2 (nuclear erythroid 2-related factor 2), is the key regulator of the response to oxidation. Rangasamy et al 53 demonstrated that Nrf2-deficient mice have heightened susceptibility to asthma and airway hyper-responsiveness. RSV infection in children leads to the rapid generation of reactive oxygen species associated with lung damage.54 Additionally, RSV could impair the Nrf2/ARE pathway to impair a host’s defence against RSV.55 MS4A3 (membrane spanning 4-domains A3; HTM4) maps to the chromosomal locus 11q13.1, which is close to the FcɛRI antigen receptor β chain gene.56 A polymorphism of MS4A3 is strongly associated with asthma.57 An association between EPB41L3 (erythrocyte membrane protein band 4.1 like 3) and RSV has not been reported, but EPB41L3 was found to be associated with in utero tobacco smoke exposure and childhood asthma.58 Although more functional studies are needed to investigate the details of pathogenesis, we found multiple genes that may be potential markers of asthma exacerbations.
We used the CAMP population to validate our genetic main effects on asthma exacerbations; however, only rs7251960 (CEACAM3) showed a significant association in CAMP (p<0.05, table 3). Differences in race/ethnicity and definition of asthma exacerbations between TCCAS and CAMP may explain our results. Moreover, RSV status is not known for subjects in CAMP. However, the LD plots of CEACAM3 from Han Chinese in Beijing, Southern Han Chinese and Ad Mixed American are similar (online supplementary figure S3). Additionally, we used a two-step integrative method combing gene expression levels and SNPs. We identified that rs7251960 is an eQTL for CEACAM3 (figure 4A-C) and is predicted to bind with several transcription factors to regulate CEACAM3 (online supplementary table S7 and S8). Although the sample size of nasal mucosa specimens was small in GSE19190 and GSE46171, we identified a reduced level of CEACAM3 mRNA expression in nasal mucosa on subjects with asthma exacerbations (figure 4D,E). Although further studies would be needed to identify additional SNP by RSV interactions in airway epithelial cells, CEACAM3 potentially modulates RSV infection status to induce asthma exacerbations.
To our knowledge, this is the first study to examine gene-by-RSV infection interactions on asthma exacerbations with a two-step integrative approach on a transcriptome-wide scale. The separation of the GEO datasets from the gene-environment study obviates the problem of statistical dependence. Our combination of several genome-wide gene expression profiles from human peripheral blood increased the statistical power, yielding novel and robust biological insights. Although we mixed healthy controls and subjects in the recovery phase as the reference group, our findings were similar when we excluded subjects in the recovery phase from the control group. While the peripheral blood response may reflect a leakage of immune cells originally activated by RSV in the respiratory tract, the response could also represent a circulating subset of immune cells involved in disease pathogenesis. One study observed that distributions of immune cells and related genes were similar in whole blood and lung.18 The immune dysregulation induced by RSV was also shown to persist beyond the acute disease.18 Therefore, an analysis of peripheral whole blood would provide an easy way to understand genomic pathogenesis by RSV latent infection. In the second step of investigating the genetic main effects on asthma exacerbations, we applied a gene-based association approach and validated in an independent cohort to reduce the probability of false positive findings. A gene-based test statistic was obtained with permutations while accounting for LD structure.37 The gene-based test offers biological inference from aggregating the information of variations in protein-coding and adjacent regulatory regions. Although we did not investigate how these SNPs influence gene or protein levels, SNPs located in the intron regions of these genes might affect the function of downstream protein products.
Despite the strengths of our study, several limitations deserve mention. First, the levels of RSV-specific IgM in serum might not constitute a good biomarker for identifying children with or without RSV infection; PCR analysis of nasopharyngeal specimens may be superior. However, a specific IgM response to RSV in children has been reported to develop soon after infection.59 Secondary infection was accompanied by accelerated antibody responses in IgM and IgG. Compared with IgG, RSV IgM antibody titres were not persistently high and would decline after an episode of infection. Therefore, in our study, RSV IgM was accepted as an immune response by RSV latent infection.60 The prevalence of exacerbations is high in both populations as the definition of asthma exacerbations was based on subject-reported episodes of nocturnal wheezing or speech-limiting severe wheezing. Nevertheless, studies have shown that nocturnal asthma is associated with future asthma-related events, such as emergency department visits, hospitalisations, oral steroid bursts and missed days of school.11 12 61 Moreover, we found that asthma exacerbations in TCCAS were significantly associated with albuterol use in the past 4 weeks, hospitalisations in the past 2 weeks, emergency department visits in the past 2 weeks and uncontrolled asthma (data not shown).
In conclusion, using our two-step integrative approach, we found that CEACAM3 modulates RSV latent infection to induce asthma exacerbations in children. Additional functional studies are necessary to investigate the roles of other genes in determining genetic susceptibility. Identification of gene–RSV interactions on asthma exacerbations may lead to new and comprehensive insights into the pathogenesis and treatment of asthma exacerbations.
Acknowledgments
The authors would like to thank the clinical assistants and pediatricians who supported data collection and all of the parents and children who participated in this study. We would also like to thank the National Center for Genome Medicine for the technical support.
References
Footnotes
Twitter @Asthma3Ways
Contributors C-HT designed the study, performed the respiratory syncytial virus (RSV) experiments, analysed the data and wrote the manuscript. ACW provided the Childhood Asthma Management Program (CAMP) data, interpreted the data and revised the manuscript. B-LC, Y-HY and S-PH provided patients’ materials and advice about data collection. M-WS performed the SNP imputation of Taiwanese Consortium of Childhood Asthma Study (TCCAS) and interpreted the data. Y-JC provided advice about the RSV experiments and interpretation of the data. YLL conceived and initiated the project, provided advice about interpretation of the data and revised the manuscript.
Funding This study was supported by grant 106-2314-B-002-131-MY3 (PI: YLL) from the Taiwan Ministry of Science and Technology, grant UN107-002, UN108-007, UN109-012 (PI: YLL and B-LC) and 107-CGN03 (PI: Y-HY and S-PH) from the National Taiwan University Hospital, grant NHRI-EX107-10606PI (PI: YLL) from the National Health Research Institutes, grant AS-TM-108-01-03 (PI: YLL) from Academia Sinica, and R01 HD085993-02 (PI: ACW) from the National Institute of Child Health and Human Development.
Disclaimer The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. Data may be obtained from a third party and are not publicly available. Data availability statement: gene expression profiles are available in the Gene Expression Omnibus resource (https://www.ncbi.nlm.nih.gov/geo/). Expression quantitative trait loci of whole blood and lung tissue are available from the Genotype-Tissue expression dataset (https://gtexportal.org/home/). The datasets supporting the conclusions of this article are included in this published article and its additional files. The raw data from the TCCAS are not publicly available. The genotype and phenotype data of the CAMP are publicly available on dbGAP.