Article Text

Abstract
Background Antibiotic resistance is a major global threat. We hypothesised that the chronic obstructive pulmonary disease (COPD) airway is a reservoir of antimicrobial resistance genes (ARGs) that associate with microbiome-specific COPD subgroups.
Objective To determine the resistance gene profiles in respiratory samples from COPD patients and healthy volunteers.
Methods Quantitative PCR targeting 279 specific ARGs was used to profile the resistomes in sputum from subjects with COPD at stable, exacerbation and recovery visits (n=55; COPD-BEAT study), healthy controls with (n=7) or without (n=22) exposure to antibiotics in the preceding 12 months (EXCEED study) and in bronchial brush samples from COPD (n=8) and healthy controls (n=7) (EvA study).
Results ARG mean (SEM) prevalence was greater in stable COPD samples (35.2 (1.6)) than in healthy controls (27.6 (1.7); p=0.004) and correlated with total bacterial abundance (r2=0.23; p<0.001). Prevalence of ARG positive signals in individuals was not related to COPD symptoms, lung function or their changes at exacerbation. In the COPD subgroups designated High γProteobacteria and High Firmicutes, ARG prevalence was not different at stable state but significantly declined from stable through exacerbation to recovery in the former (p=0.011) without changes in total bacterial abundance. The ARG patterns were similar in COPD versus health, COPD microbiome-subgroups and between sputum and bronchoscopic samples independent of antibiotic exposure in the last 12 months.
Conclusions ARGs are highly prevalent in sputum, broadly in proportion to bacterial abundance in both healthy and COPD subjects. Thus, COPD appears to be an ARG reservoir due to high levels of bacterial colonisation.
- COPD exacerbations
- bacterial infection
- infection control
Statistics from Altmetric.com
Key messages
What is the key question?
Are chronic obstructive pulmonary disease (COPD) airways an important reservoir selecting for the accumulation of antimicrobial resistance genes (ARGs) and does the resistome impact on disease?
What is the bottom line?
ARGs were common in airway samples from COPD and healthy individuals and, while they were more prevalent in the former, there was no evidence either for their selection independently from bacterial abundance, or that resistance was associated with disease severity.
Why read on?
Characterisation of the airway resistome revealed ARG prevalence dependent on bacterial burden in all those studied here, together with many features undetected by culture-based bacteriology and we suggest that further investigations of this sort could inform therapeutic practice and antimicrobial stewardship.
Introduction
The increasing prevalence of antimicrobial resistance genes (ARGs) in bacteria detected in clinical samples is a global threat1 encompassed conceptually in the term ‘resistome’.2 While the gut microbiota have been extensively studied in this regard,3 4 there is little known about the respiratory bacterial community as a reservoir of ARGs in health and disease.
Antimicrobial resistance is a particular concern in chronic obstructive pulmonary disease (COPD) and other chronic respiratory conditions where these agents are recommended for both acute and prophylactic treatment.5 While resistance may present a challenge to management, there is clear potential for such patients to become a reservoir for the dissemination of ARGs to the wider community.6 It will be important to characterise the nature and extent of ARGs in chronic lung disease to develop appropriate antimicrobial stewardship responses to this threat.
We and others have sought to better understand the course of COPD and the nature of exacerbations through microbiome analyses applied to sputum samples.7–10 In a well-studied local COPD cohort11 (designated BEAT), we specifically targeted γProteobacteria class and Firmicutes phylum using quantitative PCR and showed that the ratio between γProteobacteria and Firmicutes (γP:F ratio) in sputum samples identified an apparently antibiotic responsive subgroup of exacerbations associated with an increase in γP:F ratio at exacerbation which returned to baseline on recovery.12 This subgroup was designated High γProteobacteria, while the numerically dominant High Firmicutes subgroup showed no significant disturbance in γP:F through exacerbation to recovery. We have explored the relationships of these subgroups to ARG prevalence here.
To investigate our primary hypothesis that COPD patients constitute an important reservoir for ARGs, we undertook high-throughput quantitative PCR targeting specific ARGs in bacterial DNA extracts from sputum samples obtained from the BEAT cohort and compared these to extracts from induced sputum samples from broadly age matched health volunteers (EXCEED study). Use of serial BEAT samples extending through exacerbation events allowed us also to investigate dynamic changes in ARG levels occurring at exacerbation and any specific relationships to the High γProteobacteria and High Firmicutes subgroups. ARG profiling of bronchial brush samples from additional COPD patients (EvA study) allowed us to explore ARG prevalence in samples free from contamination by the oropharyngeal microbiota. To our knowledge we report here the first airway ‘resistomics’ study to explore and classify the type, prevalence and abundance of ARGs present in the respiratory tract in COPD and in health.
Materials and methods
Subjects and samples
Airway samples were obtained from three independent studies for which the design and endpoints have been described previously.11 13 14 In the BEAT (Biomarkers to Target Antibiotic and Systemic Corticosteroid Therapy in COPD Exacerbations) and EXCEED (Extended Cohort for E-health, Environment and DNA) studies subjects were recruited in Leicester while the EvA (Emphysema versus Airway disease)was a multicentre European study. BEAT sample sets were from consecutive stable, exacerbation onset (prior to antibiotics) and 6 weeks post exacerbation onset (recovery), visits and were available from 55 subjects. The EXCEED study is a regional genetic epidemiology study consented for recall based on genotype and phenotype. Sputum samples from 29 healthy controls were obtained; seven subjects gave a history of antimicrobial exposure (any reason) within 12 months. For the EvA subjects DNA extracts from bronchial brush samples were available from eight subjects with COPD and seven healthy controls.
DNA extraction from sputum and bronchial brush samples
For the BEAT and EXCEED studies QIAamp DNA Mini Kit (‘Gram positive bacteria extraction method’; Qiagen, California, USA) was used to extract the DNA from the homogenised sputum. For the bronchial brush samples (EvA) the Qiagen ‘AllPrep DNA/RNA Mini Kit’ was used to extract DNA and total RNA following the manufacturer’s protocol. Molecular grade water (Sigma) was used for negative controls in each extraction batch. The DNA was stored at −20°C until subsequent analysis.
High-throughput quantitative PCR
The Wafergen SmartChip Real-time PCR system (WaferGen, USA) was used to perform high-throughput qPCR reactions at the Plateforme Génomique Environnementale et Humaine in Université de Rennes 1, France. A custom set of 296 validated primer pairs targeting 283 ARGs, eight transposase genes and two integron-integrase genes and the 16S rRNA were used.15 β-globin and Actin-directed primers were also included to determine the sample human DNA content.16 17 Four ARG primer pairs were continually positive in the non-template control and were therefore discarded from the analysis.
The PCR mixture and assay conditions are detailed online supplementary files. A threshold cycle (Ct) less than 31 was used as the detection limit. The rationale for the selected detection limits is explained in the online supplement along with data showing that PCR signals for genes generally encoded in the same genomic regions are correlated (online supplementary figure S1).
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Statistical analysis
Data processing was done with Microsoft Excel 2013 (Microsoft Office 2013, Microsoft, USA). Graphs and statistical analysis were performed using GraphPad Prism (V.7, San Diego, California, USA). Parametric and non-parametric data are presented as mean (SEM) and median (IQR), respectively. Correlations were determined by linear regression. Unpaired Student’s t-tests and one way analysis of variance (ANOVA) were used to compare data between groups. Likewise, the paired t-test and repeated measures analysis of variance (RM-ANOVA) were performed for within-group comparisons. False discovery rate and Bonferroni corrected p value of <0.05 was taken as statistically significant. Heatmap graphs were produced using R (V.3.5.1) with Pheatmap package V.1.0.10.
Results
The clinical characteristics of the subjects from the COPD-BEAT, EXCEED and EvA cohorts are shown in table 1. The EvA subjects were younger and had milder COPD than the COPD-BEAT subjects since the former were recruited to undergo bronchoscopy.
Overview of clinical characteristics of study subjects
We provide raw Ct results for all our targets and samples in the online supplementary files.
Sputum ARG prevalence in COPD (BEAT) and healthy volunteers (EXCEED)
Overall analysis of sputum DNA extracts representing the exacerbation episodes of 55 subjects from the BEAT study and 29 samples from the EXCEED (healthy volunteer study) revealed positive signals respectively for 211 and 115 of the 279 targets analysed. The results for stable BEAT (n=55) and EXCEED samples from subjects without (n=22) and with (n=7) prior antibiotic exposure are shown in figure 1A, B. The proportions of positive signals associated with resistance to different antibiotic classes were similar between the three groups with minor differences in the antibiotic exposed EXCEED subjects, likely reflecting the low number in this group (figure 1A). ARGs detected per subject are compared between the groups in figure 1B and show overlapping distributions. While the median ARG positive prevalence was significantly higher in the COPD-BEAT group compared with EXCEED (37 vs 27, p=0.0019) the geometric mean bacterial burdens (16S) were also significantly lower in the latter group (3.1×108 vs 7.8×107, p=0.0003).

Analysis of ARG profile in two subject groups: COPD stable stage (BEAT) and age matched healthy controls with/without prescribed antibiotic (EXCEED). (A) Proportion of detected positive signals related to antibiotic classes affected. Numbers at column heads denote total number of positive signals. (B) Total number of ARGs detected per subject (lower Y-axis) (median±IQR) and the total bacterial load (upper Y-axis) (geometric mean with geometric SD). ARG, antimicrobialresistance gene; COPD, chronic obstructive pulmonary disease; FCA, fluroquinolone, quinolone, florfenicol, chloramphenicol, and amphenicol resistance genes; MLSB, Macrolide-Lincosamide-Streptogramin B resistance.
Heatmaps showing the prevalence of positive signals for ARGs related to agents regularly used in the management of COPD exacerbations (β-lactams, macrolides and tetracyclines) and their abundances (Ct values) for the subjects’ stable stage samples are shown in figure 2A. While strong positive signals are apparent in all four groups, clustering does not separate EXCEED from BEAT samples on the basis of the specific targets quantified, particularly after normalisation to bacterial burden (figure 2B).
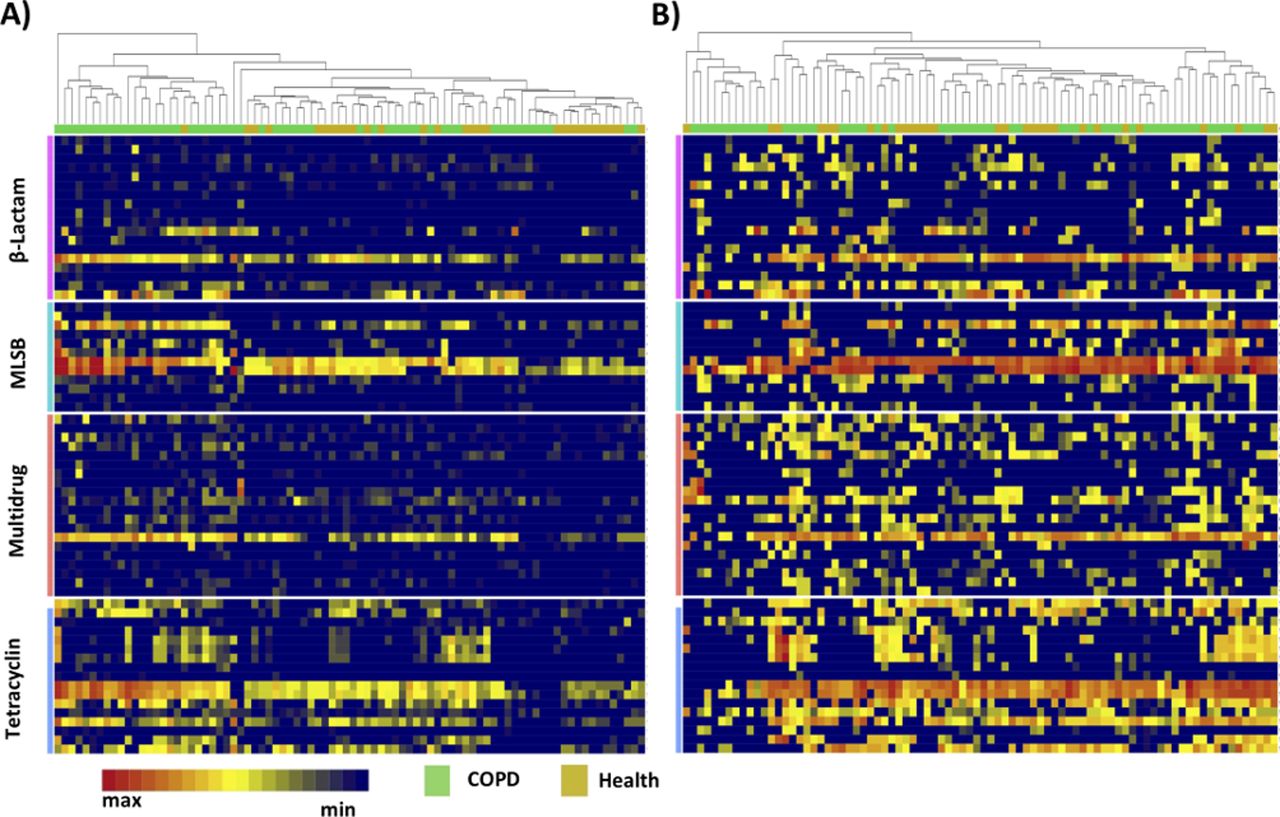
Heatmap analysis of ARG profile in two subject groups: COPD stable stage (BEAT) and age matched healthy controls with/without prescribed antibiotic (EXCEED). (A and B) Heatmaps showing prevalence and Ct values (abundance) of ARGs in samples taken at stable COPD (BEAT) and health (EXCEED). (A) Not normalised to 16SrRNA. (B) Normalised to 16SrRNA. Rows represent individual ARGs and columns show individual samples. Identities of ARGs and their resistance families are shown in online supplementary table S2. Only ARGs found in more than 90% of samples are included. ARG, antimicrobialresistance gene; COPD, chronic obstructive pulmonary disease; Ct, threshold cycle; MLSB, Macrolide-Lincosamide-Streptogramin B resistance.
We next compared ARGs detected in the COPD subgroups (BEAT) characterised by High γProteobacteria (n=20) and High Firmicutes (n=35) sputum microbiomes (online supplementary figure S2). ARG prevalence as well as the distribution of detected ARGs across different antimicrobial targets was similar in the samples from both subgroups, although a decline in prevalence was observed through exacerbation and recovery in the High γProteobacteria but not the High Firmicutes subgroup (p=0.011).
Associations between ARG prevalence and 16S signals
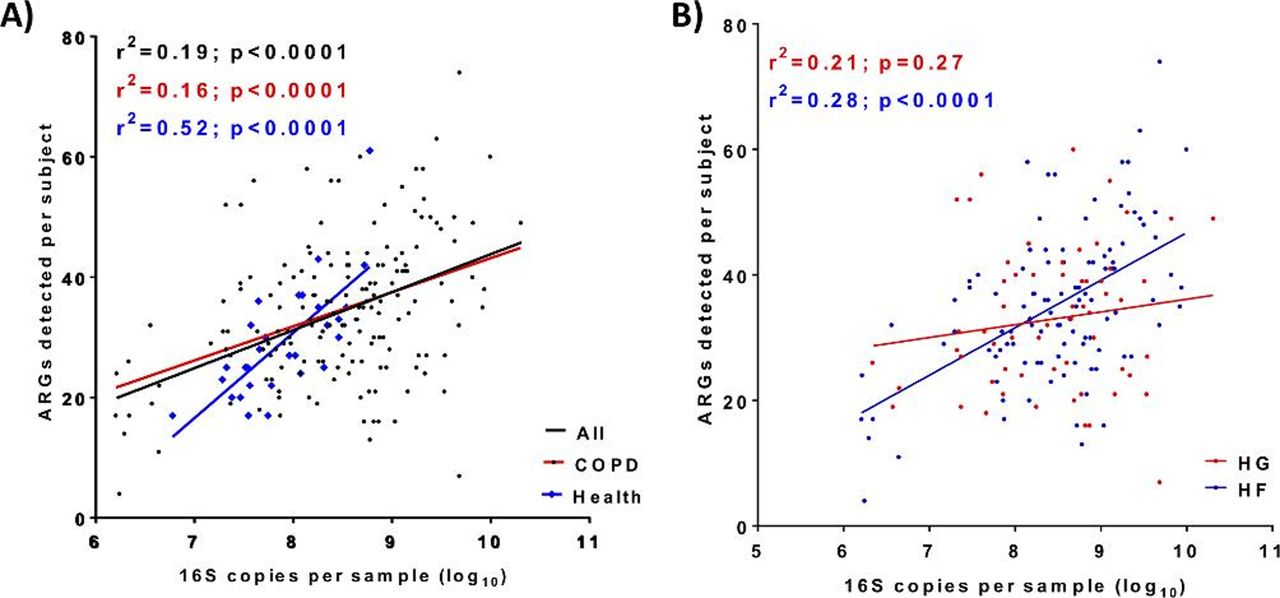
Overall ARG prevalence per sample was significantly (p<0.0001) correlated (r2=0.19) with bacterial burden (figure 3A) with a greater increment per increase in bacterial burden associated with healthy subjects. The similar analysis on the COPD-BEAT subgroups revealed that, while the High Firmicutes group showed significant correlation, the High γProteobacteria group did not, both across all samples and individually in the stable, exacerbation and recovery samples (figure 3B).

Prevalence of ARGs per sample correlates with bacterial abundance in (A) COPD (BEAT) and healthy (EXCEED) subjects. (B) COPD (BEAT) High γProteobacteria(HG) and High Firmicutes(HF) subgroups. Colours match those of the cognate data set; A) (black: all samples linear regression; red: COPD (BEAT); blue: health (EXCEED); (B) red: High γProteobacteria (HG); blue: High Firmicutes (HF). ARG, antimicrobialresistance gene; COPD, chronic obstructive pulmonary disease.
Exacerbation-associated changes in the BEAT group
To determine whether COPD exacerbations were associated with ARG prevalence, we tested for correlations with the clinical metadata collected previously.11 No significant correlations were found apart from change with FEV1 (forced expiratory volume in one second) and mCRQ (Chronic Respiratory Disease Questionnaire score) between exacerbation and recovery samples in patients receiving β-lactams (p<0.05; online supplementary table S1).
Effect of antibiotic treatment on ARG prevalence
Treatment with antibiotics is generally recognised to enrich bacterial resistance.18 However, review of our ARG prevalence data in relation to the treatment given (co-amoxiclav, amoxicillin or doxycycline) did not reveal any clear evidence that this occurred in BEAT patients (figure 4 and online supplementary figure S3). While significant variation across the three sample times was seen for several comparisons between the agent given and signals for specific ARGs, there was no consistent evidence of selection for particular determinants. This was also the case for the quantitative signals for each ARG in our BEAT samples (ie, specific ARGS were neither consistently enriched nor diminished in association with specific treatments—raw Ct data in supplement).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The number of positive reactions for each individual ARG across all subjects based on prescribed antibiotics (A–D). Non-parametric repeated analysis of variance (Friedman test) was used to compare stable, exacerbation and recovery in chronic obstructive pulmonary disease samples. ARG, antimicrobialresistance gene.
Association of ARGs with bacterial genera
We looked for correlations between ARG signals and the proportion of particular genera in the microbiome analyses previously performed on these samples10; only the 30 most abundant genera were included (>1% of reads across all samples; see table 2). Taking a false discovery cut-off of q≤0.01, it was reassuring to see strong correlations between Pseudomonas-associated sequences and the efflux protein encoding mexE and mexF genes. Less expected was the association between this genus and VanB, an ARG associated with vancomycin resistance in enterococci. Pbp2x, which is associated with increased penicillin minimum inhibitory concentrations showed the expected association with Streptococcus. Overall, eight positive genus-ARG associations were found with four genera. Three negative associations were also noted but these all failed to meet our false discovery threshold.
Genera showing significant associations with specific ARGs
Bronchial brush (EvA) sample analyses
Sputum samples are subject to contamination with the microbiota in the oropharynx. We were provided with eukaryote targeted DNA extracts from bronchial brush samples taken from eight stable COPD patients and seven healthy volunteers and these yielded 57 and 37 positive ARG signals, respectively. These results reflect lower extraction efficiency (~10-fold) and some species bias with the eukaryotic DNA extraction protocol and we characterise this in the online supplement (online supplementary figure S4). Notwithstanding this lower efficiency, the pattern of results obtained was broadly similar to those described for the BEAT and EXCEED groups (online supplementary figure S5). Despite the low sample numbers, the ARG:16S relationship was significant in the control group and, as before, demonstrated a steeper gradient than the COPD group (online supplementary figure S5).
Comparison of specific ARG prevalence across the three studies and subgroups
Table 3 shows a comparison between the per cent prevalence of positive ARG signals relevant to different antimicrobials in all three cohorts. The table is ordered according to the prevalence found in the EXCEED group. The 10 most frequently detected ARGs are almost identical in the three cohorts. Two targets, intI1 and pbp2x, were detected at least 20% more frequently in BEAT over EXCEED samples. Conversely, four targets, IS613, tet(32), pncA and ceoA were found in similar excess in the EXCEED samples.
Frequency of positive signals for highly prevalent ARGs for all three study groups
The detection frequencies (0%–38%) of targets generally considered to be of particular clinical concern (extended spectrum β-lactamases (ESBLs), AmpC β-lactamases, carbapenemases, meticillin resistance (mecA) and proteins associated with vancomycin resistance) are shown in table 4. For comparison, results for the narrow spectrum β-lactamases detected by the broad specificity blaTEM primers are included; these detect much of the TEM family from which many ESBL enzymes are derived. This target was prevalent in healthy subjects, modestly more so in COPD sputum and bronchial brush samples but was not detected in control samples from the EvA study. Overall there was a modest excess of positive signals for ESBL-encoding ARGs in the COPD samples. Primers directed to the AmpC class of generally inducible β-lactamases again showed a modest excess in BEAT over EXCEED samples although the ampC-04 directed primers showed an excess in the latter group. MecA and vanB were exclusively detected in BEAT samples. Several carbapenemase-encoding ARGs, including NDM1 (one low positive), were also detected.
Prevalences (%) of positive signals in each group for ARGs with particular clinical significance
Discussion
Antibiotics are commonly prescribed for COPD exacerbations. We therefore hypothesised that the sputum microbiota in subjects with COPD would constitute an important reservoir for antimicrobial resistance-encoding genes. With a view to enabling evidence-based antimicrobial stewardship, we have undertaken the first substantial survey of resistomes present in sputum from COPD and healthy control subjects.
ARGs were 37% more prevalent in sputum samples from COPD than those from healthy volunteers. However, this excess was predominantly attributable to the bacterial burden in sputum. Thus, COPD patients only appear to constitute an ARG reservoir inasmuch as their sputum bacterial burden is higher than that found in healthy individuals; the most prevalent ARGs were common across the sample sets. Linear regression analysis showed that for every tenfold increase in 16S signal, detection of six additional ARGs could be expected in COPD patients, while the equivalent figure would be 14 for healthy volunteers (figure 3). Perhaps, as bacterial communities in sputum approach saturation, capacity to accommodate more ARGs is somehow limited.
We next interrogated data from the 55 separate COPD exacerbation events in our samples from the BEAT study for relationships between our ARG results and our exacerbation-related metadata. Apart from modest evidence that improvements in FEV1 and mCRQ between exacerbation and recovery samples were associated with reductions in ARG prevalence, we found no clear relationships with patient clinical status. Comparison between our High γProteobacteria and High Firmicutes subgroups revealed that, while the distribution of ARG determinants was similar, prevalence declined through the exacerbations in the former but not the latter (online supplementary figure S2). We note the increase in γP:F ratio at exacerbation and return to stable levels in the High γProteobacteria subgroup compared with its stability in the High Firmicutes subgroup and suggest, as previously, that the differences observed may reflect efficacy of antimicrobial treatment in patients with an High γProteobacteria profile. Although the exacerbation frequency in the year prior to the event analysed here was greater in the High Firmicutes group (4 vs 2.2, ; ANOVA p=0.01,12), there was no clear evidence that this was due to differences in ARG profile. Nonetheless, the lack of correlation between ARG prevalence and 16S signals in the High γProteobacteria group is intriguing and adds further weight to the view that these subgroups are biologically and clinically distinct.
We found no evidence that antibiotic administration enriched ARG prevalence through exacerbations. A possible explanation for this is that such effects may be short lived and that this was missed through limiting our analyses to a 6-week post exacerbation sputum sample. There is good evidence that repeated use of antibiotics in COPD does increase the prevalence of phenotypic resistance.6
We do not know whether the ARGs detected were expressed in our samples nor do we know their bacterial hosts. However, correlation between the ARG signals and genera detected in microbiome analyses of the same samples, revealed expected correlations with Pseudomonas and Streptococcus. The Pseudomonas-VanB association is surprising since this ARG has only been reported in enterococci. This and other unexpected associations presumably reflect prior co-selection of the identified genus and low frequencies of the organisms actually carrying the ARGs.
The prevalence of pbp2x signals, which were more frequent in the BEAT samples, indicates potential for pneumococcal penicillin resistance. However, the related altered penicillin binding protein has been extensively detected in Streptococcus mitis, which is generally regarded as non-pathogenic in the lung. Nonetheless, there is clear potential for the naturally transformable S. pneumoniae to acquire pbp2x from this source.
We investigated a small number (15) of bronchial brush samples from the EvA study here as an initial assessment of the degree to which upper airway contamination may have contributed to our sputum results. Although the bacterial DNA yield was suboptimal and biassed against Gram-positive organisms, the most frequently detected ARGs were similar to those found in sputum and there were no robust differences between the COPD and control samples. A larger sample set comparing brush and sputum samples subjected to identical analytical procedures would be required to securely differentiate upper and lower airway sources of the ARG positive signals in sputum. Nonetheless, inspection of the known ARG associations of the most frequently detected genera in the human oral microbiome19 revealed that all of the detected ARGs could have come from this source.
Finally, we reviewed ARG signals relating to target genes considered sufficiently important to warrant detection of their presence or related phenotypes in most clinical microbiology laboratories. Here, the evidence for differential occurrence in COPD was stronger, with modest to low frequencies of AmpC-encoding, ESBL-encoding, carbapenemase-encoding, mecA-encoding and vanB-encoding genes detected. Two carbapenemase, three ESBLs and five AmpC genes were also detected in the EXCEED control group. Patients from whom recognised pathogens expressing these ARGs are detected in screening or clinical specimens, would often be managed by isolation. We have recently reported evidence that ARGs detected in sputum here (ermB and mefA) can also be detected in face-mask collected exhalations from COPD patients.20 While such airborne dissemination may explain the prevalence of ARG detection in our healthy volunteers, further studies will be needed to identify the risk that ARGs with high potential clinical impact may spread this way.
We were not able to find any links between the ARG signals detected here and our clinical metadata for the BEAT subjects. It is likely that both expression of antimicrobial modifying or degrading enzymes by ‘non-pathogens’ and abundance of these organisms will reduce the bioavailability and impact of antimicrobials on key pathogens. Further study will be needed to assess this possibility.
In this first survey of ARGs in the respiratory tract microbiome, several limitations must be taken into account. Positive ARG signals were assigned on the basis of a precautionary PCR Ct value in line with previous studies in this field (see online supplementary methods) and, in the absence of ARG standards, quantitation was based on differences in Ct. Given the scale of the work (223 samples), we have focused our analysis on multiple positive detections in place of confirmatory assays to assess rare positives. It could also be argued that we may have missed acute changes in the ARG profile as our exacerbation and recovery samples were taken before (day 0) and after (day 42) antimicrobial therapy. However, our main aim was to observe sustained changes.
While many of the ARGs targeted here are known to be encoded on mobile elements, many also reflect intrinsic resistance native to the host bacterial genome. Assessment of the threat of ARG dissemination from the respiratory microbiome would require a different approach from that taken here and could potentially be enabled by metagenomic analyses. Finally, the primer set deployed here has been established for several years. Numerous polymorphisms have been detected in the gene families concerned as well as recognition of novel ARGs and our coverage cannot be considered comprehensive.
While the potential challenges posed by the bacterial resistome in the lung21 22 and other body sites23 have been discussed, we are not aware of work directly comparable to the present study. Some previous respiratory studies addressing the resistome have concentrated on samples from cystic fibrosis patients24 25 and have attempted to reconcile detected resistance genes and phenotypic resistance in recognised pathogens as well as the intriguing potential of bacteriophages as vehicles of ARG dissemination.25 26 Most resistome studies have focused on faecal samples to assess the gut reservoir of ARGs and relationships to interchange with the local environment and geographic variations thereof.27–34 While the general airborne microbiome may be a source of the ARGs we detected here, we have recently observed ARGs in exhaled breath from human subjects20 and these potential sources require further investigation. Transit between body sites must also be considered and we note with interest recent application of the concepts of island biogeography in this context.35 As recently reviewed,36 the key challenge remains translating human resistome analyses into clinical benefit.
In conclusion, we have found that while the human respiratory tract is a reservoir for numerous ARGs, their prevalence in samples appears more dependent on bacterial load than clinical condition; healthy subjects with similar ages to our COPD patients carried most of the same resistance determinants. Key further questions include understanding the relative contributions of antibiotic and environmental exposure to prevalence of ARGs and their influence on clinical outcomes.
Supplemental material
Acknowledgments
The authors thank all the research volunteers who participated in this study.
References
Footnotes
CB and MRB are joint senior authors.
Contributors MRB, CB and MRO designed the study, MYR and VM delivered the ARG-related laboratory analyses. Samples and analyses and analyses were contributed by KH and MB (BEAT), LG, CJ, NFR and MT (EXCEED), and LZ-H, IG (EvA). RCF was responsible for data management. MYR, CEB, and MRB drafted the manuscript and all authors approved the final version.
Funding This work was funded in part by Airway Disease Predicting Outcomes through Patient Specific Computational Modelling (AirPROM) project (funded through FP7 EU grant), Emphysema versus Airways Disease (EvA FP7; 200506), Wellcome Trust Senior Fellowship (MT), Medical Research Council (COPD-BEAT), National Institute for Health Research (NIHR) Leicester Respiratory Biomedical Centre. This paper presents independent research funded by the NIHR. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Competing interests None declared.
Patient consent for publication All subjects provided written informed consent.
Ethics approval Ethics approvals were obtained for each study (08/ H0406/189, 13/EM/0226, 08/H0402/19).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Linked Articles
- Airwaves